产地上海
可售地**
品牌SUNGO
型号SUNGO BV
包装纸质
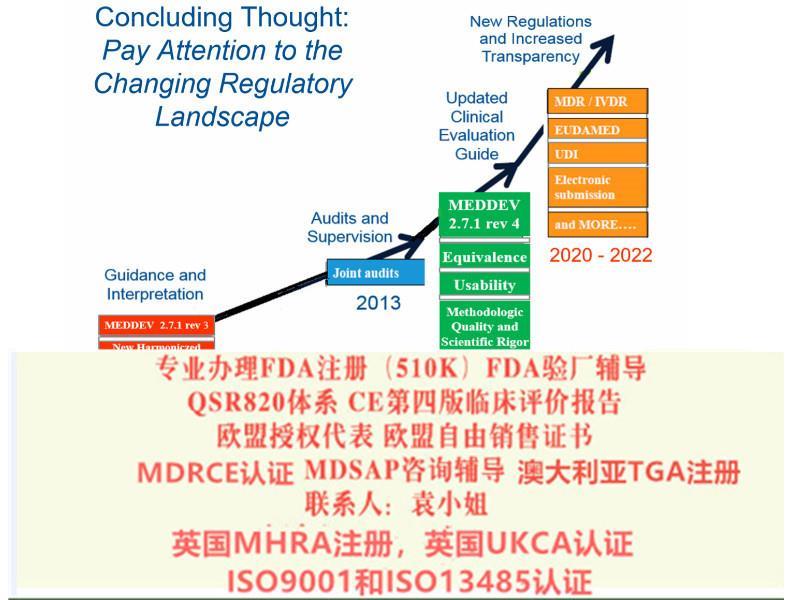
MDR还包括在互联网上销售器械以及用于远程提供诊断或服务的器械(第6条)。MDR为一些Ilb类器械和植入性Il类器械引入了由立小组进行的评估咨询程序(第54条)。
ISO 17664的规定适用于用于侵入性或其他直接或间接接触患者的设备。ISO 17664中没有定义处理指令,相反,ISO 17664规定了一些要求,以协助器械制造商提供详细的加工说明,包括下列活动(如适用):
a) 使用时的初始处理;
b) 清洗前准备;
c) 清洁;
d) ;
e) 干燥;
f) 检查和维修;
g) 包装;
h)
i) 存储
j) 运输
需要办理以下认证可以随时找我 :
1:出口欧盟:MDR CE认证/IVDRCE认证,欧盟授权代表,欧盟注册,欧盟自由销售证书
2:出口英国:英国代表,英国MHRA注册,UKCA认证,英国自由销售证书
3:出口美国:美国FDA注册,FDA510K,QSR820体系
4:中国:国内的器械注册证和生产许可证
5:出口加拿大:加拿大的MDEL注册
6:质量管理体系认证:ISO13485咨询和认证

根据欧洲法规的要求,制造商应起草符合性声明,对于加贴CE标志上市的产品的符合性负有责任。因此,制造商在产品进入欧洲市场前,需要评估并选定适用的指令和符合性路
径,确保产品符合法规要求,然后加贴CE标志。其主要过程包括:
1. 确定根据指令的定义,产品是否属于器械,哪个欧盟指令适用于所考虑的器械:器械指令(93/42 / EEC),体外诊断器械指令(98/79/EC)或有源植入式器械指令(90/385 / EEC)
2. 确定器械的分类。
3. 实施质量管理体系(如适用)。大多数公司使用ISO 13485来满足要求。
4. 准备CE标记的技术文件(Technical Documentation)或设计文档(Design Dossier)。
5. 如果制造商在欧洲没有实际的场地,需要任命一位欧洲授权代表,代表制造商在欧盟境内进行相关的法规联络事宜。
6. 制造商的质量管理体系(QMS)和技术文件/设计文档由公告机构(Notified Body)审核和评估。若器械属于I类非、I类无测量功能或顾客定制产品,则不需要公告机构的介入。
7. 从公告机构处获得CE证书和ISO 13485证书。
8. 起草符合性声明(DoC),其中所涉及的器械符合相应的指令。
9. 在产品上(包括标签、说明书等)加贴CE标志,并投放欧洲市场。对部分欧洲,可能需要在上市前在该国器械主管当局处进行备案。
10. 维持质量体系和CE证书的有效性。

并可在2020 年5 月26 日前并通知符合新法规的符合性评估机构。公告机构可在2020 年5 月26 日前, 采用合规的符合性评估流程并按照新法规签发证书。对于特定Ⅲ类器械和Ⅱ b 类器械产品,在已委派必要的器械协调小组(MDCG)、小组前提下,同样可通过指令豁免在2020 年5 月26 日之前投放市场。法规关于公告机构的要求(正文第35~50 条) 自2017 年11 月26日起适用,即公告机构在新法规发布后的六个月内即应开始进行相应的申请,符合要求后方可依据新法规开展符合性评估。同时法规对成员国主管机构的和MDCG 的成立也设定了期限,要求于2017 年11 月26 日前完成。对于成员国主管机构之间的协调,设定期限为2018 年5 月26 日。

市场 (MDR第93~99条)
主管机构对器械的符合性特性和性能进行适当检查,包括酌情审查文件以及基于适当样品的物理或实验室检查。确保器械符合相关欧盟的相关要求,并且不危害健康、或公共利益保护的任何其他方面。
主管机构应制定年度活动计划,并分配足够数量的胜任人力和物质资源以执行这些活动。
我公司办理:ISO13485认证,CE评估报告编写 等产品出口的相关认证
http://sungofda.cn.b2b168.com