儿童体温贴MDR的CE第四版临床评价报告 CE技术文件 申请要求
发货地址:上海市金山区
产品数量:9999.00个
价格:面议
周期4周
品牌SUNGO
公司SUNGO
流程SUNGO
国家欧洲
MDD指令和MDR法规的CE认证的区别
老MDD指令申请CE认证,由于法规规定产品在市场上出现任何问题,都是由制造商承担。其中欧盟授权代表的职责只是沟通协调以及产品包装可以使用欧盟授权代表的公司名字和地址信息的责任。
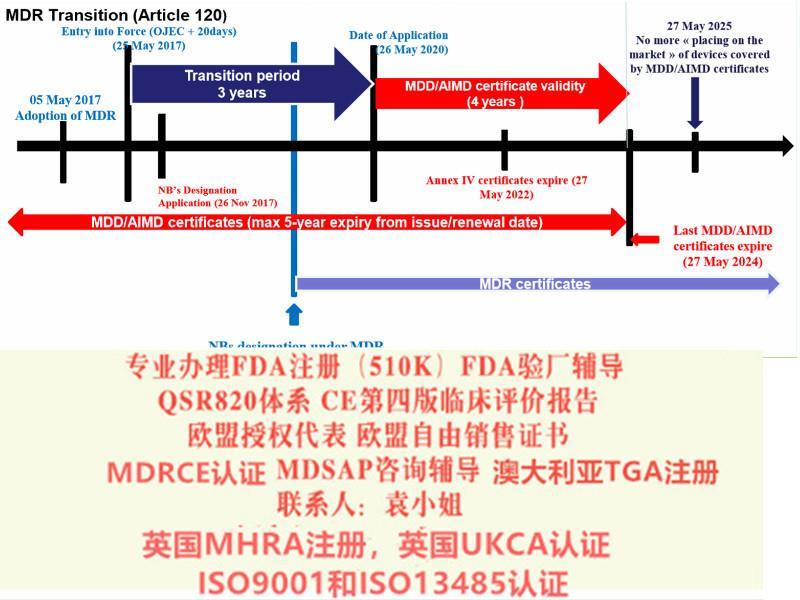
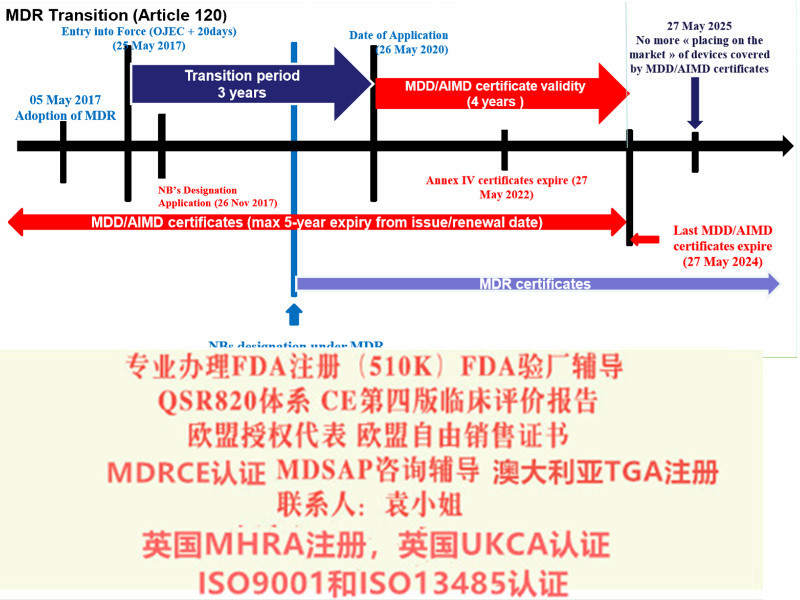
欧盟第四版评价(MEDDEV 2.7.1 Rev 4)指南主要变化a)报告更新的频率b)报告编写人和评价人的c)评价报告需要有明确的可测量的目标d)确定技术发展水平e)数据的科学性和有效性f)比对器械g)比对器械的数据获得h)什么时候需要试验i)风险-收益j)售后监督和售后跟踪8)提出Eudamed数据库的建立和使用9)提出器械的可追溯性(UDI)10)对NB提出严格的要求2017年5月5日,欧盟(Official Journal of the European Union)正式发布了欧盟器械法规(REGULATION (EU) 2017/745,简称“MDR”)。MDR将取代Directives 90/385/EEC (有源植入类器械指令)and 93/42/EEC(器械指令)。
MDR对于DOC的要求 MDR 在其附录 IV中对DOC的内容作了明确的规定,至少包括如下内容: 制造商名称、注册商品名或注册商标和单一注册号(如签发)及其授权欧盟代表(如适用)和注册营业地点的联系地址 制造商对签发欧盟符合性声明负完全责任的声明 附录VI第C部分所所述的基本的器械标识UDI - DI

清洗验证ISO 17664 ISO 17664适用于拟由用户或准备使用的第三方处理的设备的制造商。这包括用于重复使用和需要处理的设备,使其从使用后的状态变为清洁、和/或状态,并为下次使用做好准备一次性器械,供应时未经、但在清洁、和/或无菌状态下使用,因此在使用前需要进行处理。
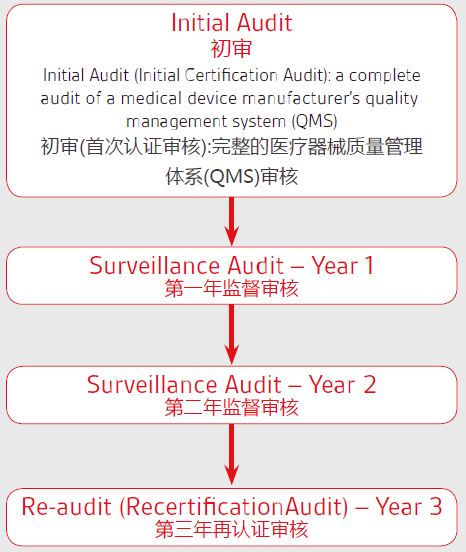
我们的咨询业务 1:MDR法规培训 新法规立法过程、变化及转换期 MDR覆盖的范围,包括和MDD, AIMD的修订要点及主要区别 MDR法规结构及条款清单 MDR分类规则要求 MDR对经销相关方 (Economic Operators)要求 MDR符合性审核程序 质量管理体系的全新要求,以及MDR与ISO 13485:2016的关系 通用和性能要求GSPR MDR对技术文档TCF的要求 评价CER上市后追踪PMCF的要求 上市后监督PMS的要求 MDR中对器械性标识UDI要求; 欧盟符合性声明 (EU declaration of conformity) 要求 器械欧盟数据库(European Database on Medical Devices, EUDAMED)介绍及输入 公告机构的审核准则 充分准备以应对MDR欧盟器械法规相关的审核 Q&A 2:专项,MDR法规变化-UDI和标签 MDR法规变化-GSPR MDR法规变化-PMS、PMCF系统 MDR法规变化-质量管理体系 MDR法规变化-技术文件 MDR法规变化-评估,调查 MDR法规变化-风险管理 可用性评价 3.欧代和注册服务SUNGO 荷兰和德国公司可以提供欧盟授权代表服务,同时提供向当地部门申报注册的服务。 4.整体MDR升级换版实施服务 包含上述1、2、3的全部内容,还包括针对公告机构审核开具的不符合的整改服务。 我们的服务流程 1 预评估:简要管理,以确保清楚了解MDR的重要性和业务影响 预评估考虑组织的挑战:管理意识,人员配备能力和可用性,预算影响 2.差异分析 评估对产品、内部资源、组织和预算的影响 检查新的分类规则(MDR I, IIa,IIb,III类),确认现有和产品的符合性评估路线 核对器械定义,确认是否属于扩大范围或属于附录16中所涉及的属于器械的范围 检查产品与关NB机构有关的要求 审查现有技术文档(技术文件)的变更 评估及更新质量管理体系(以下第3点) 检查可用证据和风险管理的充分性,识别差异(第56条) 评估产品标签(附件I第Il1章) 确保上市后监督的安排充分适宜够(第七章第1节) 制定上市后性能跟踪(PMCF,附件XIV B部分) 做好迎接新的警戒需求的规定(第七章第二节) 确保可追溯相关方面的义务(第3章) 3,质量体系评估 评估新IVDR法规下QMS符合标准和流程的程度 增加新法规应用于QMS的要求 协助和识别合规负责人(PPRC)并参与培训 4,公告机构NB确认 选择合适的公告机构,确认公告机构资质及范围 建立新法规实施过渡计划 5,技术文件编制 编制符合MDR要求的技术文件(TD) 编制评估报告、生物学评价报告和风险管理等技术文件等 产品设计开发流程,确保输入及输出的完整性 确认标签、上市后监督、上市后性能跟踪方案 技术文件整改(风险管理报告,性能评估报告,GSPR等) 6,QMS建立:更新现有体系中IVDR用于QMS的要求 定制企业合规QMS系统 执行体系实施计划确保覆盖各个方面及各方面责任 7, 可追溯性UDI 建立可追溯性QMS要求 建立UDI系统程序及制度 确认UDI的规划及实施
SUNGO公司介绍:SUNGO创建于2006年。以助力大健康产品**流通为使命,我们致力于成为受用户信赖的合规服务机构。SUNGO的客户覆盖**洲,遍布30多个和地区,客户总数**过5000家。、、中国器械企业**过30%选择SUNGO,同时也有多家**器械企业选择SUNGO提供服务。
http://sungofda.cn.b2b168.com