随着历史的发展,ISO组织在此基础上又将此标准进行了修订,升级为ISO13485:2016
顾客为了确保得到的产品长期稳定地满足质量要求,纷纷要求供方按ISO9000族标准建立质量体系并通过认证,要求供方用健全的质量体系来保证供货产品满足要求且质量稳定,因为单纯靠成品抽样检验不可能有稳定的产品质量
ISO 13485:2016 Medical devices——Quality management systems——Requirements for regulatory (医疗器械 质量管理体系 用于法规的要求),是2017年11月为止的现行版本。
我公司专业办理医疗产品出口欧盟、美国、中东南美等国家的各种认证:
TUV莱茵,TUV南德,***等CE认证,全套CE技术文件编订, CE第四版临床评价(MEDDEV 2.7.1 Rev 4)编写,欧盟授权代表,欧盟自由销售证书,ISO13485:2016,医疗器械单一审核程序(MDSAP)审核、美国FDA注册(含FDA510K申请), FDA QSR820验厂辅导及整改,FDA警告信应对&RED LIST REMOVAL/QSR820体系辅导/OTC验厂辅导及整改,英国BRC认证咨询,BSCI验厂辅导;口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试、防护服type5/6 (PPE认证咨询(89/686/EC个人防护指令辅导)。
一、新版标准有哪些变化?
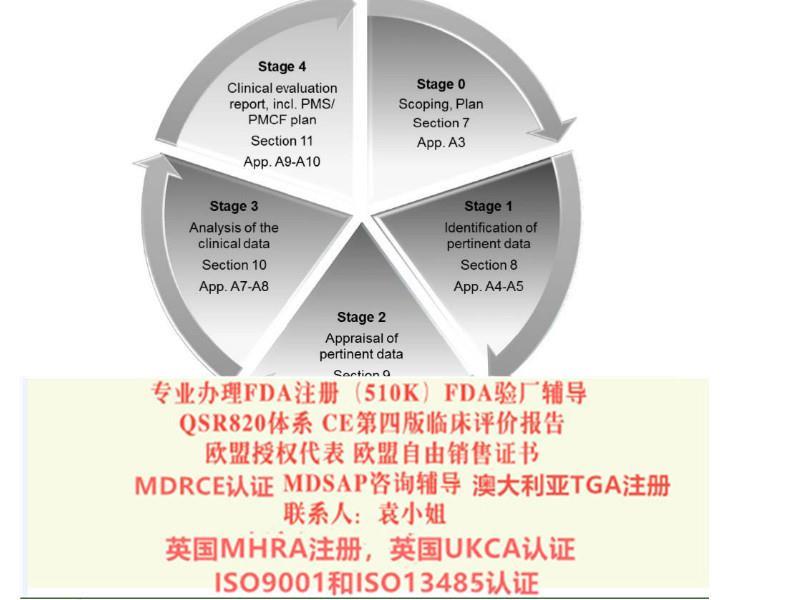
ISO13485:2016生命周期的主题已经引入,贯穿产品设计开发,生产,储存,安装和处置的每个阶段,风险管理得到加强,许多监管要求已被纳入。
以下是ISO13485:2016的主要修订和变化,可作为企业进行ISO13485:2016转换的参考:
1、范围清楚地解释到其适用于产品的存储和分配,内部和外部供应商服务,以及相关服务,例如:设备分销商,运输服务,灭菌服务,安装维护服务,软硬件配件供应商。
2删减条款更为合理。
新版延伸了不适用的条款,允许客户在有合理原因下删减第6,7或8条。这种变化符合当前的业务发展模式,反映了标准的适用性。
3、术语和定义更贴近实际。
条款已经被添加及修改。2003版的定义已经被修改和删除。新版本包括临床评估,生命周期,制造商,进口商,分销商,授权代表,绩效评估,投诉,上市后的监控,风险和风险管理等新技术术语。
4、条款变更的更为合理。
新版本更多地考虑到监管的要求,并强调需遵守监管要求,以确保医疗设备的*性和有效性。
许多监管要求已被采纳,如美国的FDAQSR820,日本的JPAL MO No.169,巴西GMP,EU-MDR和EU-IVDR以及加拿大,澳大利亚和中国的监管要求,使其具有更广泛适用性。
例如,介绍了可用性和软件应用要求; 控制设计过程更为详细; 明确规定了变更控制的要求; 加强对供应商管理的要求; 定义了可追溯性(UDI)的要求和目的; 添加了反馈和投诉处理的要求。
5.、风险管理的要求得到加强,新版本更加明确。
许多地方都提到“风险”和“风险管理”。提出对供应商风险的控制。医疗器械风险,反馈机制,投诉处理和数据分析更为详细,从而使得上市后的风险监控系统更加可操作。

在现代贸易实践中,,第二方审核早就成为惯例,又逐渐发现其存在很大的弊端:一个供方通常要为许多需方供货,第二方审核无疑会给供方带来沉重的负担;另一方面,需方也需支付相当的费用,同时还要考虑派出或雇佣人员的经验和水平问题,否则,花了费用也达不到预期的目的。唯有ISO9000认证可以排除这样的弊端。因为作为**方的生产企业申请了第三方的ISO9000认证并获得了认证证书以后,众多第二方就不必要再对**方进行审核,这样,不管是对**方还是对第二方都可以节省很多精力或费用。还有,如果企业在获得了ISO9000认证之后,再申请UL、CE等产品品质认证,还可以免除认证机构对企业的品质保证体系进行重复认证的开支。
2017年11月为止的执行版本是ISO13485:2016《医疗器械质量管理体系用于法规的要求》
-/gjhdff/-
http://sungofda.cn.b2b168.com