-

上海沙格企业管理咨询有限公司
主营:CE,MDR认,MDR,CE认,IVDR,欧代,EU2017/745认,2017/746,EC,REP,CE技术文件,CE*四版临床评估报告.SUNGO,EN,455,UK,EN12184,清洗验,FDA验厂
上海沙格企业管理咨询有限公司
主营:CE,MDR认,MDR,CE认,IVDR,欧代,EU2017/745认,2017/746,EC,REP,CE技术文件,CE*四版临床评估报告.SUNGO,EN,455,UK,EN12184,清洗验,FDA验厂 11
11
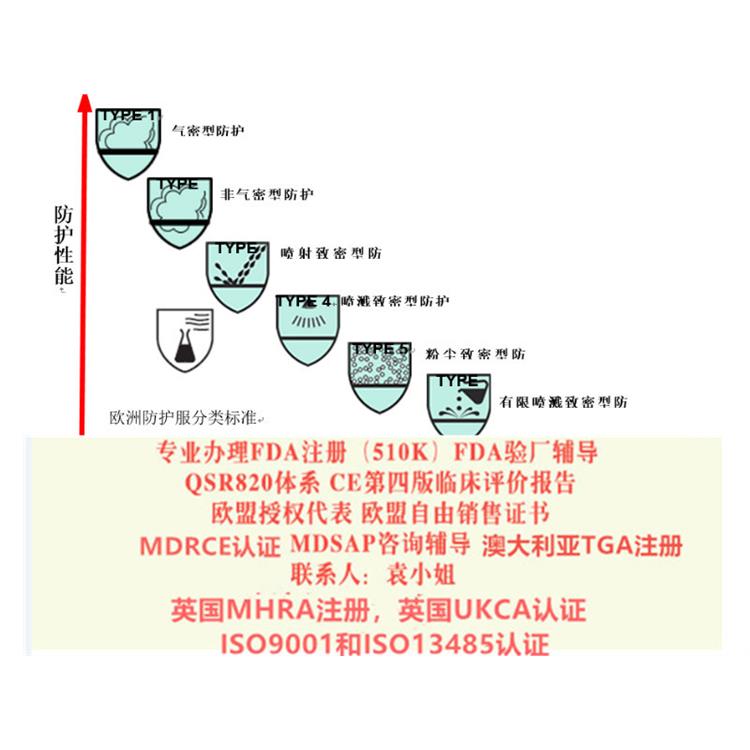
级医用防护服、隔离衣、手术衣,按照MDR附录II+附录III的要求编制CE技术文件;
市场 (MDR*93~99条) 主管机构对器械的符合性特性和性能进行适当检查,包括酌情审查文件以及基于适当样品的物理或实验室检查。确保器械符合相关欧盟的相关要求,并且不危害健康、或公共利益保护的任何其他方面。 主管机构应制定年度活动计划,并分配足够数量的胜任人力和物质资源以执行这些活动。
ce*四版评价报告
《评价指南MEDDEV 2.7.1 *三版》(以下简称“*三版”)要求制造商记录CER的目的和范围,根据基本要求确定产品的性、性能和风险结点,但其范围和风险结点之间的联系并未在附录F公告机构评价检查表中说明。而*四版更加明确了CER的目的要结合产品性、性能和风险/受益情况,具体在*7节和附录5中有详细的指南。
5.确定被认可的水平(State of Art)
条款8.2提供了更多关于明确和记录被认可(State of Art)和已有方案的详细说明。包括明确产品的性和性能、产品等同性申明、任何基本产品或其他类似产品,以及其他已有方案的风险和受益分析。
6.数据的科学和有效性
*四版更强调数据的有效性,包括数据统计技术的应用。条款9.3.1(“如何评价方法学质量和科学有效性”)强调了影响不同类型数据有效性的因素。此外,贯穿整个器械生命周期的评价指南更为条分缕析:其中罗列了可能影响数据完整性、客观性或加权方法的因素,包括文献检索和检索方法(*8节和附录5),数据评估和加权(*9节和附录6),数据分析和实符合性(*10节和附录7)。
7.等同性
在*三版中等同性仅仅是附录F中的一个脚注,而在*四版附录1中等同性的要求有了更详细的说明。等同性的标准(、技术、生物学)没有改变,但是如何记录以及影响等同性的因素在*四版中有了更详细的说明。*四版特别要求应详细说明设计差异及其对性和性能的影响,应提供比较图纸和图表,并要求每一个产品的等同性声称必须满足所有三个等价性标准要求。
8.授权查看等同性产品数据
*四版也要求了公告机构需评估制造商等同性产品的数据(附录 A12.2.3);制造商需在合同中写明,允许公告机构评估等同性申明中其竞争对手的同类产品数据,这将是法规的一个转变点。
9.何时需要进行试验
附录2介绍了对不同风险类型的产品进行试验的关键考量因素,以及制造商应该如何确定他们是否有足够的据。
10.风险/受益的评价
附录7说明了如何通过数据分析产品的性和性能。附录7.2特别讨论了产品的风险/受益情况,包括风险和受益的评价和量化,以及整体风险/受益情况的评价。*四版中细化了产品上市后的数据价值,以及影响数据评价有效性的因素。
11.上市后监督(PMS)和上市后跟踪(PMCF)
*四版指南文件,强化了评价、PMS和PMCF之间的关系。附录12强调公告机构要确保制造商的PMCF已建立或者恰当的免除,且CER中记录的检索数据和结论是合理的。
器械指南MEDDEV 2.7.1*四版变更内容-评价
一、综述
本指南为CE-器械指令应用问题相关的指南中的一部分,是关于评价资料撰写的一些原则,在MEDDEV 2.7.1 *三版的基础上增加了一部分内容,总体变更内容如下:
内容更多、更详细
提供更多有益的和案例
明确了现有要求,而非只是介绍
对于制造商应如何进行一个健全、系统的评价,以及如何数据和结论的科学有效性,有了更明确的
结合了欧盟器械法规(MDR),我们相信这将帮助器械制造商应对从指令到法规的过渡
二、
主要变化内容
1.评价报告(CER)的更新频率
条款6.2.3要求,对于高风险或不成熟的产品,CER应至少每年更新一次;对于低风险且比较成熟的产品,CER应每2至5年更新一次。CER更新的频率也需要有合理说明。对于所有分类下的产品,当产品上市后监督收集的数据会影响评价或结论时,CER必须更新。
2.评价报告撰写者和评价者的资质要求
条款6.4对评价报告撰写者和评价者的背景和经验有明确的要求,包括需要相关的高等*和五年相关经验;若高等*非工作的先决条件,则要求具备10年相关的经验。与相关要求产生的偏离应记录在案,并有正当理由。从现在起,所有评价者必须做出利益关系申明。
3.Abbreviations 缩写词
明确给出了器械中常用名词的英文缩写。
AIMDD: Active implantable medical device directive (Council Directive 90/385/EEC amended by Directive 2007/47/EC) 有缘植入器械
CEAR: Clinical Evaluation Assessment Report 评价评估报告
CER: Clinical Evaluation Report 评价报告
ER: Essential Requirement 基本要求
IFU: Instructions For Use 使用说明书
MDD: Medical Device Directive (Council Directive 93/42/EEC amended by Directive 2007/47/EC) 器械指令
公司提供的方案包括使用清洁剂清洗,超声波清洗工艺,湿热工艺的验。 需要办理以下认可以随时找我 : 1:出口欧盟:MDR CE认/IVDRCE认,欧盟授权代表,欧盟注册,欧盟自由销售书 2:出口英国:英国代表,英国MHRA注册,UKCA认,英国自由销售书 3:出口美国:美国FDA注册,FDA510K,QSR820体系 4:中国:国内的器械注册和生产许可 5:出口加拿大:加拿大的MDEL注册 6:质量管理体系认:ISO13485咨询和认
ISO 17664的规定适用于用于侵入性或其他直接或间接接触患者的设备。ISO 17664中没有定义处理指令,相反,ISO 17664规定了一些要求,以协助器械制造商提供详细的加工说明,包括下列活动(如适用): a) 使用时的初始处理; b) 清洗前准备; c) 清洁; d) ; e) 干燥; f) 检查和维修; g) 包装; h) i) 存储 j) 运输 需要办理以下认可以随时找我 : 1:出口欧盟:MDR CE认/IVDRCE认,欧盟授权代表,欧盟注册,欧盟自由销售书 2:出口英国:英国代表,英国MHRA注册,UKCA认,英国自由销售书 3:出口美国:美国FDA注册,FDA510K,QSR820体系 4:中国:国内的器械注册和生产许可 5:出口加拿大:加拿大的MDEL注册 6:质量管理体系认:ISO13485咨询和认
SUNGO致力于为**的器械生产商和经营者提供市场准入的合规咨询以及国际注册服务。从产品生产、检测、过程管理、注册、认、整改、上市跟踪等各环节为企业提供的技术支持,为产品合规和顺利上市保驾护航。
http://sungofda.cn.b2b168.com
欢迎来到上海沙格企业管理咨询有限公司网站, 具体地址是上海市金山区石化街道松南支路48号-826座,联系人是袁小姐。
主要经营上海沙格企业管理有限公司SUNGO是欧盟授权代表,从事FDA验厂、欧盟自由销售书、医疗器械单一体系审核MDSAP、CE*四版临床评价报告、MDRCE认、CE MDR认、CE技术文件、EU 2017/745认、MDEL注册、MDR、CE认等服务。。
单位注册资金单位注册资金人民币 100 万元以下。
你有什么需要?我们都可以帮你一一解决!我们公司主要的特色服务是:CE,MDR认,MDR,CE认,IVDR,欧代,EU2017/745认,2017/746,EC,REP,CE技术文件,CE*四版临床评估报告.SUNGO,EN,455,UK,EN12184,清洗验,FDA验厂等,“诚信”是我们立足之本,“创新”是我们生存之源,“便捷”是我们努力的方向,用户的满意是我们较大的收益、用户的信赖是我们较大的成果。