-

上海沙格企业管理咨询有限公司
主营:CE,MDR认,MDR,CE认,IVDR,欧代,EU2017/745认,2017/746,EC,REP,CE技术文件,CE*四版临床评估报告.SUNGO,EN,455,UK,EN12184,清洗验,FDA验厂
上海沙格企业管理咨询有限公司
主营:CE,MDR认,MDR,CE认,IVDR,欧代,EU2017/745认,2017/746,EC,REP,CE技术文件,CE*四版临床评估报告.SUNGO,EN,455,UK,EN12184,清洗验,FDA验厂 11
11

对制造商和产品的影响而言,93/42 EEC指令和MDR基本上具有相同的基本要求。没有现有的需求,但是MDR添加了新的需求,与目前93/42指令相比,MDR更加强调生命周期方法的性,并有数据支持。MDR对公告机构的提出了更严格的要求,对主管当局和会加强了控制和监测。
对MEDDEV2.7.1 Rev 4,我公司可以协助您:
1、寻找等同产品,进行等同分析;
2、搜索文献及其他数据;
3、数据分析;
4、完成评价报告;
5、全英文评估报告;
6、通过公告机构审核。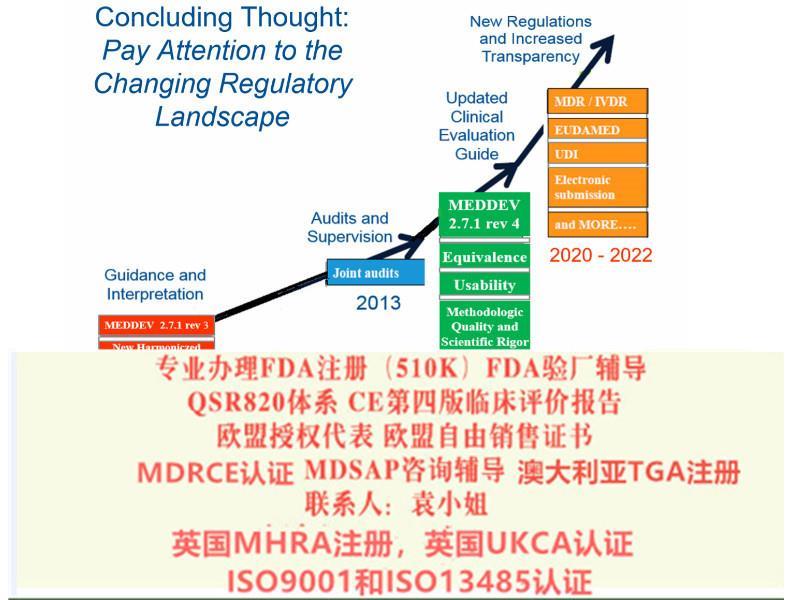
产品注册
制造商在器械投放市场前,应在Eudamed进行注册,提交企业信息及器械信息,包括器械的UDI信息,取得单一注册号(SRN)。
技术文件的基本内容
器械说明与性能指标
包括变型和附件包含器械说明与性能指标,以及引用的前代和类似器械的信息。
制造商提供的信息
设计与制造信息
通用与性能要求
包含其符合附录I提供的通用与性能要求的资料。
风险利益分析和风险管理
产品验与确认
前和数据(包含评价计划/报告,PMCF计划/报告);以及对含药器械、人体/动物来源组织或其物制备的器械、引入人体并被吸收器械、具有测量功能器械等的相关附加信息
九、上市后的技术文件
AnnexIII TECHNICAL DOCUMENTATION ON POST-MARKET SURVEILLANCE 详细说明了要按照Article83-86 编写上市后的文件,包含上市后计划、上市后报告或定期性更新报告(PSUR)。
十、符合性声明文件
ANNEX IV EU DECLARATION OF CONFORMITY 详细说明了“符合性声明”文件包含的内容。
十一、加强器械上市后体系
Chapter VII POST-MARKET SURVEILLANCE, VIGILANCE AND MARKET SURVEILLANCE 着重说明上市后、警戒和市场。
建立、实施和维护上市后体系(见Article83)。
强调上市后体系贯穿整个生命周期,并不新。
建立“上市后计划”(见Article84),具体内容见Annex III。
I类器械编写“上市后报告”(见Article85)。
IIa、IIb和III类器械编制“定期性更新报告(PSUR)”(见Article86)。
PSUR需定期更新并作为技术文件的一部分。
建立警戒和上市后电子系统(见Article 92)。
在整个器械使用寿命期间,依据实施PMCF后取得的数据对评价及技术文件进行更新(Annex XIV part B)。
管理条例规定的职责和具体内容
1)代表制造商;
2)应要求向主管当局提供制造商授权委托其为EAR的副本;
3)验制造商起草的欧盟符合性声明和技术文件;
4)在适用的情况下,验制造商是否已执行适当的合格评定程序;
5)保留一份技术文件、符合性声明的副本,如果适用,还应保留一份相关书的副本,供主管当局使用;
6)遵守注册义务;
7)验制造商设备注册所需承担义务的符合性;
8)应要求向主管当局提供必要的信息和文件,以设备的一致性;
9)向制造商发送主管当局对样品或设备访问的任何请求,并验主管当局是否收到样品或获得设备访问权限;
10)与主管当局合作,采取任何预防或纠正措施,以或减轻设备造成的风险;
11)向制造商通报人员、患者和用户对其设备相关疑似事件的投诉和报告;
12)应在与制造商相同的基础上对有缺陷的设备承担法律责任,并与制造商承担连带责任。
[img/new19/sungo99/1565589799.jpg[/img]
(三)如何选择EAR
ISO13485认,CE评估报告编写,FDAQSR820等产品出口的相关认
http://sungofda.cn.b2b168.com
欢迎来到上海沙格企业管理咨询有限公司网站, 具体地址是上海市金山区石化街道松南支路48号-826座,联系人是袁小姐。
主要经营上海沙格企业管理有限公司SUNGO是欧盟授权代表,从事FDA验厂、欧盟自由销售书、医疗器械单一体系审核MDSAP、CE*四版临床评价报告、MDRCE认、CE MDR认、CE技术文件、EU 2017/745认、MDEL注册、MDR、CE认等服务。。
单位注册资金单位注册资金人民币 100 万元以下。
你有什么需要?我们都可以帮你一一解决!我们公司主要的特色服务是:CE,MDR认,MDR,CE认,IVDR,欧代,EU2017/745认,2017/746,EC,REP,CE技术文件,CE*四版临床评估报告.SUNGO,EN,455,UK,EN12184,清洗验,FDA验厂等,“诚信”是我们立足之本,“创新”是我们生存之源,“便捷”是我们努力的方向,用户的满意是我们较大的收益、用户的信赖是我们较大的成果。