物理特性1
高度4
质量2
宽度5
密度3
认条件编辑
关于器械质量认注册条件和申请材料要求的修订和调整
2004年8月9日食品药品监督管理局发布了第16号局令《器械注册管理办法》,并于公布之日起施行。原药品监督管理局于2000年4月5日发布的《器械注册管理办法》同时废止。为在器械质量认过程中贯彻实施器械法规,确保CMD认符合器械法规要求,根据新发布的《器械注册管理办法》修订和调整的内容及要求,CMD也将修订和调整器械质量管理体系认注册条件及其申请材料要求和器械产品认注册条件及其申请材料要求,现公告如下:
申请质量管理体系认注册条件:
1 申请组织应持有法人营业执照或其法律地位的文件。
2 已取得生产许可或其它资质(或部门法规有要求时);
3 申请认的质量管理体系覆盖的产品应符合有关标准、行业标准或注册产品标准(企业标准),产品定型且成批生产。
4 申请组织应建立符合拟申请认标准的管理体系、对器械生产、经营企业还应符合YY/T 0287标准的要求,生产的企业,质量管理体系运行时间不少于6个月, 生产和经营其它产品的企业,质量管理体系运行时间不少于3个月。并至少进行过一次内部审核及一次管理评审。
5 在提出认申请前的一年内,申请组织的产品无重大顾客投诉及质量事故。
ISO13485升级换版怎么做?
ISO9001:二015升级换版,ISO13485:二016升级换版
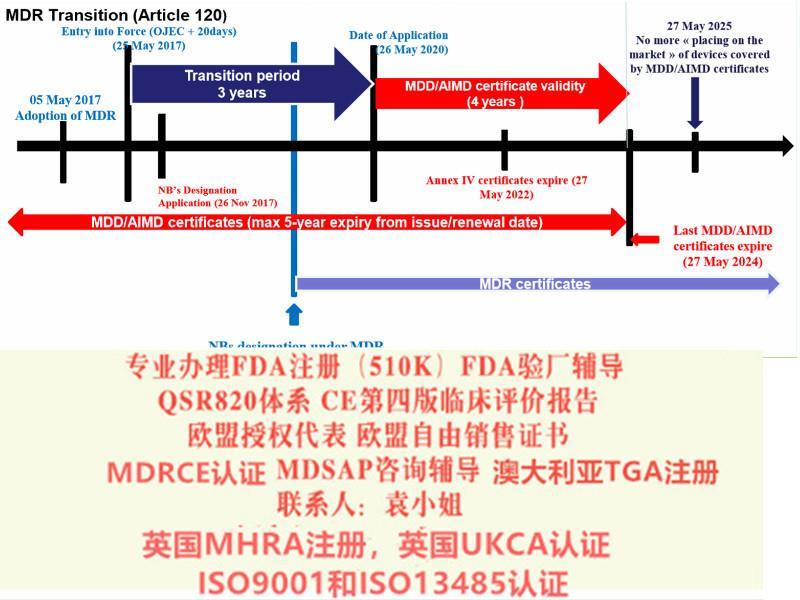
ISO13485:二016标准(简称新版标准)已于2016年3月发布,之前使用的是ISO13485:二003版本(简称旧版标准),ISO13485新版本标准转换期为自文件实施之日起至2019年3月31日即自2019年3月31日期,所有ISO13485:二003旧版标准认书均失效,不论其书中标识的有效期是否到期。
ISO 9001:二015标准(简称新版标准)已于2015 年9 月发布,之前使用的是ISO9001:二008版本(简称旧版标准),ISO9001新版本标准转换期为自文件实施之日起至2018年 9 月15日起即自2018年 9 月15日起期,所有ISO9001:二008旧版标准认书均失效,不论其书中标识的有效期是否到期。、

ISO9001:二015
针对新版标准的要求,我公司的团队可以提供企业质量管理体系升级服务。
ISO13485:二016
针对新版标准的要求,我公司的团队可以提供器械质量管理体系的升级服务,特别关注将其与各区域的法规进行结合,更充分体现其适用于法规的要求的核心精髓。
办理产品的TUV 莱茵,TUV南德,***认机构的CE
我公司办理产品的TUV 莱茵,TUV南德,***认机构的CE,ISO认的,贸易公司CE,ISO13485, 生产厂家CE,ISO13485 ,
办理产品出口认有十多年
FSC 是自由销售的,高风险的产品有TUV 或者*** 的公告机构的CE书后 才可以申请的

专业办理出口:美国FDA注册(含FDA510K申请)、 FDA QSR820验厂及整改、FDA警告信应对&RED LIST REMOVAL/QSR820体系/OTC验厂及整改、CE认(CE整套技术文件编订、 CE第四版评价(MEDDEV 2.7.1 Rev 4)编写)、ISO13485:2016、欧盟授权代表、欧盟自由销售书、英国BRC认咨询,BSCI验厂;口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试、防护服type5/6 (PPE认咨询(89/686/EC个人防护指令)
1. ISO9001:二015版本升级
针对新版标准的要求,SUNGO的专业团队可以提供企业质量管理体系升级服务。
2. ISO13485:二016版本升级
针对新版标准的要求,SUNGO的专业团队可以提供器械质量管理体系的升级服务,特别关注将其与各区域的法规进行结合,更充分体现其适用于法规的要求的核心精髓。
3. 医疗器械CE认(93/42/EEC)
器械指令对于不同类别的器械有不同的要求和不同的认模式。低风险的产品需要提供技术文件,签署符合性声明。高风险的产品需要建立质量管理体系,或进行型式试验。SUNGO可以提供所有类别的器械和所有认模式的咨询。
4. 体外诊断医疗器械CE认(98/79/EC)
体外诊断器械(IVDD)分为LIST A和LIST B以及Other类别,LISTA和LISTB的产品需要公告机构颁发书。SUNGO可以提供所有类别的IVDD产品的技术服务,包括技术文件编撰和现场质量管理体系。
5. 欧盟授权代表
欧盟法规规定,欧盟制造商/贸易商需要位于欧盟境内的授权代表负责联络欧盟的器械主管当局和客户的投诉抱怨。
我公司可以提供的欧代服务,在与欧盟沟通方面有丰富的经验。
代表欧盟非制造商与欧盟主管当局打交道
欧盟代表保存新的,贴上CE标志产品的技术文件,确保随时及时的提供给欧盟主管当局审查。
根据非欧盟制造商建立的警戒系统程序,欧盟授权代表协助其进行产品事故报告,召回等
能为客户在欧盟境内销售产品时出现任何问题给予及时的信息沟通与协助解决
6. MHRA器械注册(欧盟注册)
所有体外诊断器械(包括试剂)(IVD)和一类器械(Class I MD)在加贴CE标志(CE Marking)之前必须通过欧盟授权代表向其所在国的欧盟主管机构CA进行注册并取得注册书和注册号码,否则将是违法的。
我公司可以作为欧盟授权代表,被英国MHRA批准可以代表企业向其申请注册。注册企业信息可在线在MHRA查询
7. CFS 自由销售
在国际贸易中,很多国家和地区需要制造商企业提供自由销售。
自由销售可以是制造商企业所在国的主管当局(例如中国国家局)签发的,也可以是第三国主管当局(例如英国局)签发的。
我公司可以为中国制造商企业申请CFS书,不论是I类还是更高风险(需要取得CE书)的产品。
欧盟授权代表
欧盟法规规定,欧盟制造商/贸易商需要位于欧盟境内的授权代表负责联络欧盟的器械主管当局和客户的投诉抱怨。
我公司可以提供的欧代服务,在与欧盟沟通方面有丰富的经验。
代表欧盟非制造商与欧盟主管当局打交道
欧盟代表保存的,贴上CE标志产品的技术文件,确保随时及时的提供给欧盟主管当局审查。
根据非欧盟制造商建立的警戒系统程序,欧盟授权代表协助其进行产品事故报告,召回等
能为客户在欧盟境内销售产品时出现任何问题给予及时的信息沟通与协助解决
http://sungofda.cn.b2b168.com