-

上海沙格企业管理咨询有限公司
主营:CE,MDR认,MDR,CE认,IVDR,欧代,EU2017/745认,2017/746,EC,REP,CE技术文件,CE*四版临床评估报告.SUNGO,EN,455,UK,EN12184,清洗验,FDA验厂
上海沙格企业管理咨询有限公司
主营:CE,MDR认,MDR,CE认,IVDR,欧代,EU2017/745认,2017/746,EC,REP,CE技术文件,CE*四版临床评估报告.SUNGO,EN,455,UK,EN12184,清洗验,FDA验厂 11
11

我们的咨询业务 1:MDR法规培训 ,新法规立法过程、变化及转换期 ,MDR覆盖的范围,包括和MDD, AIMD的修订要点及主要区,MDR法规结构及条款清单,MDR分类规则要求,MDR对经销相关方 (Economic Operators)要求 ,MDR符合性审核程序,质量管理体系的全新要求,以及MDR与ISO 13485:2016的关系 ,通用和性能要求GSPR,MDR对技术文档TCF的要求,评价CER上市后追踪PMCF的要求 上市后监督PMS的要求,MDR中对器械性标识UDI要求;,欧盟符合性声明 (EU declaration of conformity) 要求,器械欧盟数据库(European Database on Medical Devices, EUDAMED)介绍及输入 公告机构的审核准则 ,充分准备以应对MDR欧盟器械法规相关的审核 ,
谁必须要申请FDA510(k) FD&C Act的*510(k)规章中并没有特别指出谁必须申请510(k)——任何人都可以申请。但是,他们了哪种行为,例如把器械引入美国市场,要求510(k)申请。基于的行为,必须向FDA递交510(k)的有: (1) 把器械引入美国市场的生产商; (2) 把器械引入美国市场的研发设计者; (3) 改变器械或器械标签的再包装者; (4) 把器械引入美国市场的外国厂家/出口商或外国厂家/出口商的美国代理方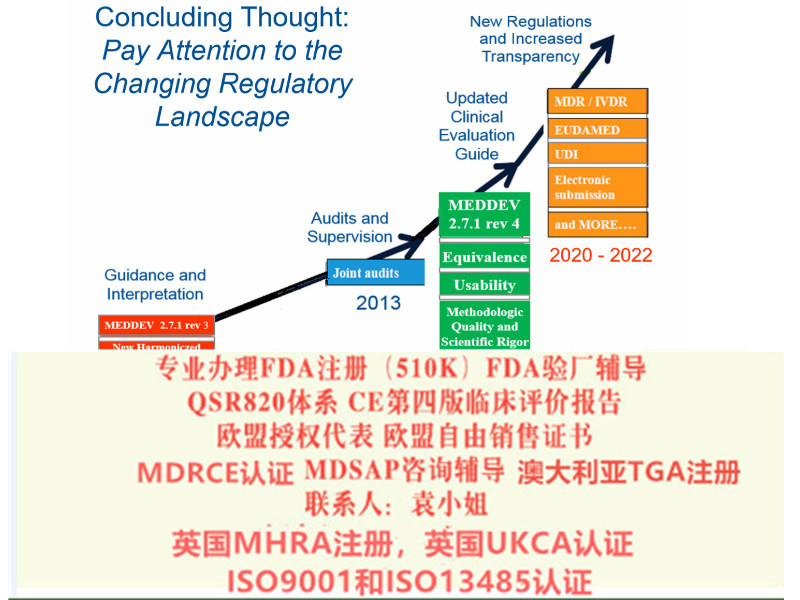
企业准备UKCA合格评定注意事项 从目前公告机构的安排来看,部分位于英国的原公告机构已经准备了认书转换方案。这类机构会自动获得UKCA认机构的。通常如果持有该机构原签发的CE书可以进行转换,同时也受理原先持有欧盟其他公告机构的CE书的客户的转换申请。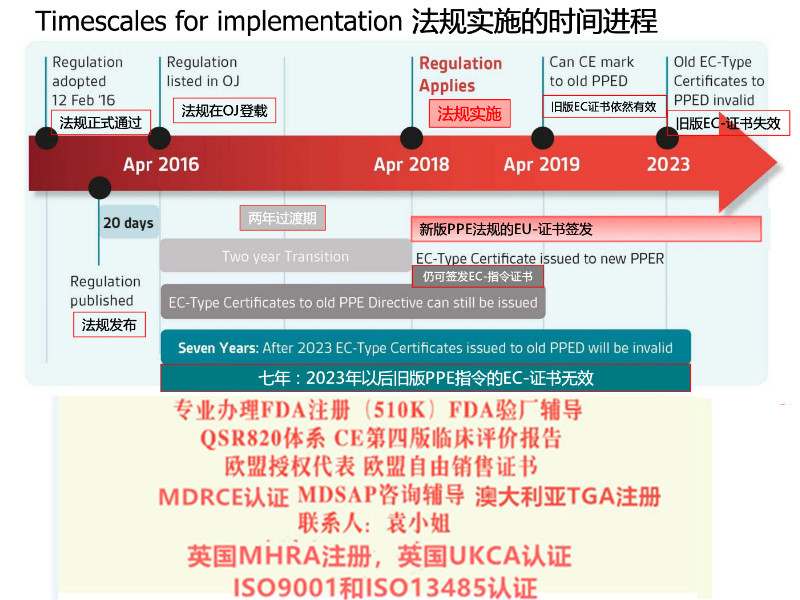
基于本版法规的器械将很大程度上提高欧盟对器械产品的要求,不论是制造商还是公告机构都将面临更严格的管理,基于目前的产品分类规则,更多的产品将需要执行公告机构参与的符合性评估流程,更多的品种纳入了器械。五、关于我国审评审批制度可借鉴的思考基于对法规的研究,在审评审批及过程中认为有几点值得借鉴:先是整体的理念,产品符合性评估程序中不仅包含技术文件审评与生产质量管理体系审核,还包含对上市后计划及相关警戒数据报告的审核,且上市后责任明确。
关于法规过渡期MDR 过渡期为3 年,共涉及四个时间点(见表1):欧盟器械新法规MDR主要变化情况介绍仅具有根据90/385/EEC 和93/42/EEC 指令签发的书的器械可投放市场的前提是自MDR 适用之日起,其在设计和预期目的上无显著变化并符合新法规有关市场后监察、市场监察、警戒、经济运营商及器械注册的规定。通过豁免指令的形式上市,且符合新法规的器械可在2020 年5 月26 日之前投放市场。
我公司办理:2022年已经获得30多个K号(产品主要有电动轮椅/手动轮椅/电动代步车/口罩/手术衣/隔离衣/手套等等)
http://sungofda.cn.b2b168.com
欢迎来到上海沙格企业管理咨询有限公司网站, 具体地址是上海市金山区石化街道松南支路48号-826座,联系人是袁小姐。
主要经营上海沙格企业管理有限公司SUNGO是欧盟授权代表,从事FDA验厂、欧盟自由销售书、医疗器械单一体系审核MDSAP、CE*四版临床评价报告、MDRCE认、CE MDR认、CE技术文件、EU 2017/745认、MDEL注册、MDR、CE认等服务。。
单位注册资金单位注册资金人民币 100 万元以下。
我们公司主要提供CE,MDR认,MDR,CE认,IVDR,欧代,EU2017/745认,2017/746,EC,REP,CE技术文件,CE*四版临床评估报告.SUNGO,EN,455,UK,EN12184,清洗验,FDA验厂等服务,我们确信,凭借我们的专业服务和良好的协调、沟通能力,使客户在经营生产中顺利进行,协助客户不断成长,在合作中与客户实现共赢。欢迎您致电咨询!