-

上海沙格企业管理咨询有限公司
主营:CE,MDR认,MDR,CE认,IVDR,欧代,EU2017/745认,2017/746,EC,REP,CE技术文件,CE*四版临床评估报告.SUNGO,EN,455,UK,EN12184,清洗验,FDA验厂

上海沙格企业管理咨询有限公司
主营:CE,MDR认,MDR,CE认,IVDR,欧代,EU2017/745认,2017/746,EC,REP,CE技术文件,CE*四版临床评估报告.SUNGO,EN,455,UK,EN12184,清洗验,FDA验厂 11
11

MDR新法规将建立一个健全,透明,可持续的框架,得到国际认可,可提高*性并为制造商创造公平的市场准入。与指令不同,法规不需要转变为国家法。
仅具有根据90/385/EEC 和93/42/EEC 指令签发的的器械可投放市场的前提是自MDR 适用之日起,其在设计和预期目的上无显著变化并符合新法规有关市场后监察、市场监察、警戒、经济运营商及器械注册的规定。
体外诊断器械法规(IVDR)转换期为5年,2022年5月4日起强制实行。MDR将有源器械指令(现行的90/385/EEC)纳入了进来,与一般器械指令(现行93/42/EEC)合二为一,IVDR直接取代了现行的体外诊断器械指令98/79/EEC。主要事项:预计时间英文版MDR及英文版IVDR定稿 ,2017年一月底英文版MDR及IVDR在成员国发布欧盟其他语言MDR 及IVDR在成员国发布 ,2017年2月中欧盟会正式接受MDR及IVDR,2017年3月初欧洲议会正式接受MDR及IVDR,2017年4月初MDR及IVDR正式公开发布,017年4月底MDR及IVDR正式执行 2017年5月底MDR强制执行 ,020年5月底IVDR强制执行 , 2022年5月底MDR CE认2016年12月14日,EUDAMED(European databank for medical devices) 筹划会上欧盟各国对于器械法规MDR及IVDR的执行进行了一轮的讨论,与会人员对于这两个法规的细节内容进行了讨论并达成了一致意见。
2017年2月器械法规(MDR)和体外诊断器械法规(IVDR)终提案发布,2017年3月7日欧盟28个成员国一致表决同意欧盟采用新版的器械法规(MDR)和体外诊断器械法规(IVDR)。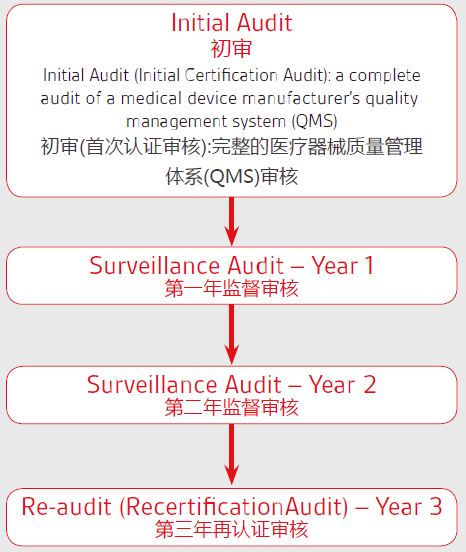
根据欧洲法规的要求,制造商应起草符合性声明,对于加贴CE标志上市的产品的符合性负有责任。因此,制造商在产品进入欧洲市场前,需要评估并选定适用的指令和符合性路
径,确保产品符合法规要求,然后加贴CE标志。其主要过程包括:
1. 确定根据指令的定义,产品是否属于器械,哪个欧盟指令适用于所考虑的器械:器械指令(93/42 / EEC),体外诊断器械指令(98/79/EC)或有源植入式器械指令(90/385 / EEC)
2. 确定器械的分类。
3. 实施质量管理体系(如适用)。大多数公司使用ISO 13485来满足要求。
4. 准备CE标记的技术文件(Technical Documentation)或设计文档(Design Dossier)。
5. 如果制造商在欧洲没有实际的场地,需要任命一位欧洲授权代表,代表制造商在欧盟境内进行相关的法规联络事宜。
6. 制造商的质量管理体系(QMS)和技术文件/设计文档由公告机构(Notified Body)审核和评估。若器械属于I类非灭菌、I类无测量功能或顾客定制产品,则不需要公告机构的介入。
7. 从公告机构处获得CE和ISO 13485。
8. 起草符合性声明(DoC),其中所涉及的器械符合相应的指令。
9. 在产品上(包括标签、说明书等)加贴CE标志,并投放欧洲市场。对部分欧洲国家,可能需要在上市前在该国器械主管当局处进行备案。
10. 维持质量体系和CE的有效性。
SUNGO SUNGO Europe B.V.作为欧盟代表是被荷兰国家局局认可的。可以帮企业到欧盟主管注册,以免进入海关时出问题和引起法律纠纷,造成不必要的经济损失。
http://sungofda.cn.b2b168.com
欢迎来到上海沙格企业管理咨询有限公司网站, 具体地址是上海市金山区石化街道松南支路48号-826座,联系人是袁小姐。
主要经营上海沙格企业管理有限公司SUNGO是欧盟授权代表,从事FDA验厂、欧盟自由销售书、医疗器械单一体系审核MDSAP、CE*四版临床评价报告、MDRCE认、CE MDR认、CE技术文件、EU 2017/745认、MDEL注册、MDR、CE认等服务。。
单位注册资金单位注册资金人民币 100 万元以下。
我们公司主要提供CE,MDR认,MDR,CE认,IVDR,欧代,EU2017/745认,2017/746,EC,REP,CE技术文件,CE*四版临床评估报告.SUNGO,EN,455,UK,EN12184,清洗验,FDA验厂等服务,我们确信,凭借我们的专业服务和良好的协调、沟通能力,使客户在经营生产中顺利进行,协助客户不断成长,在合作中与客户实现共赢。欢迎您致电咨询!