-

上海沙格企业管理咨询有限公司
主营:CE,MDR认,MDR,CE认,IVDR,欧代,EU2017/745认,2017/746,EC,REP,CE技术文件,CE*四版临床评估报告.SUNGO,EN,455,UK,EN12184,清洗验,FDA验厂

上海沙格企业管理咨询有限公司
主营:CE,MDR认,MDR,CE认,IVDR,欧代,EU2017/745认,2017/746,EC,REP,CE技术文件,CE*四版临床评估报告.SUNGO,EN,455,UK,EN12184,清洗验,FDA验厂 11
11

制造商的质量管理体系(QMS)和技术文件/设计文档由公告机构(Notified Body)审核和评估。若器械属于I类非、I类无测量功能或顾客定制产品,则不需要公告机构的介入。
IVDR IVD产品如果想进入欧盟市场,需要CE认 欧盟会(EC)的DG SANTE部门(健康和食品总局)负责这件事情 但各成员国的具体的落实需要靠CA(器械部门),由NB(备案机构)按照欧盟指令和法规要求执行器械产品的符合性评估和认 执行的标准开始是按照90年代出的器械法规中的IVDD来IVD,但是由于这个标准太松,2017年正式上了新法规IVDR,2022年取代IVDD 原来按照IVDD标准批的书2024年将过期 也是现在还处于过渡期 新的法规更加关注:产品、性能评估、评价、上市后 产品分类也有了变化:class ABCD classA的要求可能,产家自我声明即可 class BCD都需要NB 通过了CE认的产品可以在36个流通,一些东南亚也认可
我公司办理产品出口欧盟、美国、中东南美等的各种认:
TUV莱茵,TUV南德,***等CE认,CE技术文件编订, CE*四版评价(MEDDEV 2.7.1 Rev 4)编写,欧盟授权代表,欧盟自由销售书,ISO13485:二016,器械单一审核程序(MDSAP)审核、美国FDA注册(含FDA510K申请), FDA QSR820验厂及整改,FDA警告信应对&RED LIST REMOVAL/QSR820体系/OTC验厂及整改,英国BRC认咨询,BSCI验厂;口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试、防护服type5/6 (PPE认咨询(89/686/EC个人防护指令)。
CE评价报告
编订:CE*四版本评价报告(MEDDEV 2.7.1 Rev 4)
1.不变的是什么?变化的是什么?
一般原则仍然是制造商必须使用数据来器械符合相关的基本要求(Essential Requirements)。这些数据仍须基于本器械的(上市前)研究数据、其他同类器械的研究数据以及来自上市后监督(PMS)活动和警戒活动的数据。仍需对数据进行收集、评价和分析,但是,评价应该详尽到什么程度、什么评价方法是可接受的以及应该何时进行评价,这些内容几乎是全新的。
2.何时进行评价?
评价贯穿于器械的整个生命周期,包括器械的设计阶段。评价是一个持续不断的过程,应当有相应的文件记录。对于评价报告(CER)的更新频率,制造商应给出合理解释,更新频率应根据风险、科学发展、设计变更等相关因素来确定。若通过上市后监督(PMS)收集到可能改变评价的相关数据,应对评价报告进行修订。对于创新或高风险器械,其评价报告应每年更新一次。对于其它器械,其评价报告应至少每2到5年更新一次。制造商必须对更新频率做出合理解释。
3.如何进行评价?
评价的不同阶段涉及范围和计划的定义、数据识别和每个数据集的评价、数据分析、和评价报告的定稿。对于大多数制造商来说,评价并不陌生,不过新版MEDDEV 2.7.1对这些阶段进行了详细的描述。新版指南也详述了进行评价的人员资质要求。另外,对于评价的范围,新版MEDDEV 2.7.1提供了更详细的要求。同时新版指南也详述了在哪里进行文献检索、如何进行文献检索、以及如何记录这些文献的收集、评价和分析过程。
CE评价报告
4.是否仍可使用实质等同器械?
新版指南下,仍可使用实质等同器械的数据。但是“实质等同”的概念在新版指南中有了明确的定义,解读的空间相比之前要小很多。实质等同器械应当几乎是完全相同的。新版MEDDEV 2.7.1 的附录A1对实质等同性进行了详细说明。要器械的实质等同,必须考虑到器械的、技术和生物特性。新版MEDDEV 2.7.1的附录A1对这三方面的特性进行了详细说明。而且器械需要满足所有这三方面的特性才能实质等同。在此背景下,新版指南还描述了“差异”的概念,以及验此概念所需的步骤。实际上,实质等同器械的使用将局限于同一制造商的器械,而且符合要求的器械必须来自相同的器械系列。即便是同一器械的换代产品,要想实质等同,也需仔细考量。
5.评价报告(CER)长啥样?
新版MEDDEV 2.7.1的要求非常清晰,因为它不仅要求对数据进行分析,而且还要求制造商透明公开所采用的方法和步骤。评价报告的实质部分应包括一个日志,记录该评价是如何进行的。相应的,评价报告必须有附录,包括检索策略、全部检索结果、评价策略和结果、数据分析、以及清晰的参考文献列表。此外,所有文章和报告应可供审核员核实之用。
6.新的欧盟器械法规(MDR)情况如何?
提议的器械法规(MDR)的妥协统一文本已于2016年6月发布。预计在2020年的季度,新的器械法规(MDR)将正式实施。新的器械法规(MDR)要求高质量的评价。新版MEDDEV 2.7.1向此方向迈出了坚实的一步。按照新版MEDDEV 2.7.1进行评价也可帮助制造商为即将的立法做准备。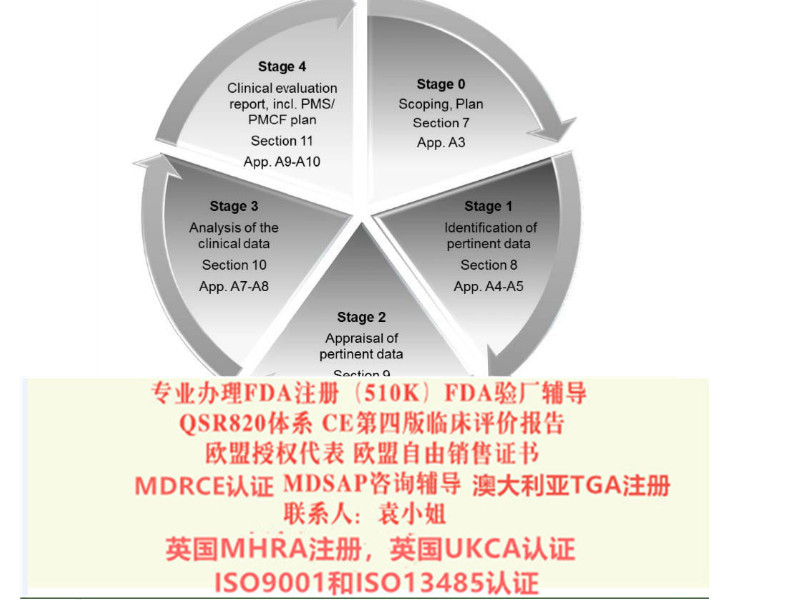
当公告机构现场审核或者文件审核开具不符合时,我们提供整改服务,确*整改验。项目背景()欧洲议会和理事会于2017 年4 月5 日签发的关于器械*2017/745 号法规, 修订了*2001/83/EC 号指令,*178/2002 号(EU)法规和*1223/2009 号(EU)法规,并废除了理事会*90/385/EEC 号和*93/42/EEC 号指令.MDR实施之后,在三年过渡期内仍然可以按照MDD和AIMDD申请CE书并保持书的有效性。依据Article 120 clause2 的规定,过渡期内NB签发的CE书继续有效。
欧盟主管部门按产品的危险程度,将产品分为:Ⅰ类、Is(),Im(测量),Ⅱa类、Ⅱb类、Ⅲ类 关于I类产品申请CE认(CE认的新法规是:2017/745 MDR新法规)的流程如下: 1:那么I类申请CE的流程是 : 企业自己必须确保自己的产品符合欧盟法规,产品是有效的,并建立相关技术文件,签署符合性声明,确定欧盟授权代表(我们提供的欧代是荷兰公司),然后到成员国主管当局注册登记(到荷兰局CIBG注册登记)获得注册信函之后,即可在产品加贴CE标志进入欧盟销售。 不需要TUV,BSI等这些Notified Body公告机构参与审核发书的 2:SUNGO提供的欧盟授权代表的职责的,不是Notified body公告机构,*到欧盟授权网站查询 3:终给企业的文件是:MDRCE技术文件+MDR 符合性声明+欧盟注册信函
我们的核心资源包括分布在**主要经济体的运营网络,具有美国IAS认可资质的实验室,具有ANAB认可资质的认机构,以及分布**的资源。
http://sungofda.cn.b2b168.com
欢迎来到上海沙格企业管理咨询有限公司网站, 具体地址是上海市金山区石化街道松南支路48号-826座,联系人是袁小姐。
主要经营上海沙格企业管理有限公司SUNGO是欧盟授权代表,从事FDA验厂、欧盟自由销售书、医疗器械单一体系审核MDSAP、CE*四版临床评价报告、MDRCE认、CE MDR认、CE技术文件、EU 2017/745认、MDEL注册、MDR、CE认等服务。。
单位注册资金单位注册资金人民币 100 万元以下。
我们公司主要提供CE,MDR认,MDR,CE认,IVDR,欧代,EU2017/745认,2017/746,EC,REP,CE技术文件,CE*四版临床评估报告.SUNGO,EN,455,UK,EN12184,清洗验,FDA验厂等服务,我们确信,凭借我们的专业服务和良好的协调、沟通能力,使客户在经营生产中顺利进行,协助客户不断成长,在合作中与客户实现共赢。欢迎您致电咨询!