-

上海沙格企业管理咨询有限公司
主营:CE,MDR认,MDR,CE认,IVDR,欧代,EU2017/745认,2017/746,EC,REP,CE技术文件,CE*四版临床评估报告.SUNGO,EN,455,UK,EN12184,清洗验,FDA验厂

上海沙格企业管理咨询有限公司
主营:CE,MDR认,MDR,CE认,IVDR,欧代,EU2017/745认,2017/746,EC,REP,CE技术文件,CE*四版临床评估报告.SUNGO,EN,455,UK,EN12184,清洗验,FDA验厂 11
11

标准变化编辑
ISO 13485:2012是欧盟使用的标准,当前国际通用的标准仍然是ISO 13485:2003版,2012版的升级相比ISO 13485:2003变化有如下几点,ISO 13485:2012年在标准的前言部分做了适当的调整,主要是用词的变更,以及一些适用范围细节的修改,在附录部分做了较大的改动,修改了ANNEX ZA, ANNEX ZB以及ANNEX ZC这三个目录,增加了ISO 13485与三个器械指令之间的关系。此次的升级2012版本值得期待,该标准仍然只是在欧盟所推行,国内仍然使用ISO 13485:2003。
强调法规要求
如,新标准5.2"以顾客为关注焦点"要求,"管理者应确保顾客的要求得到确定并予以满足",而不是"管理者应以增强顾客满意为目的,确保顾客的要求得到确定并予以满足"。
又如,新标准8.2.1的标题为"反馈",而不是"顾客满意"。这是因为顾客满意不适合作为器械行业的法规目标。
这种修改与新标准促进全世界管理体系法规的协调目标是一致的。
文件的程序、作业书
根据器械行业的特点,新标准要求形成文件的程序、作业书或要求。
1.文件控制程序(4.2.3)。
2.记录控制程序(4.2.4)。
3.培训(6.2.2)。
注:或地区法规可能要求组织建立用于识别培训需求的形成文件的程序。
4.基础设施维护(当维护活动或缺少这种维护活动可能影响产品的质量时,组织应建立形成文件的维护活动要求,包括它们的频次)。
5.工作环境(6.4)。
①当人员与产品或工作环境的接触会对产品质量有不利影响时,组织应建立对人员健康、清洁和服装的形成文件的要求;
②如果工作环境条件能对产品质量产生不利影响,组织应建立形成文件的工作环境条件要求和程序或作业书,以监视和控制这些工作环境条件;
③适当时,为了防止对其它产品、工作环境的污染或人员的影响,组织应建立对受污染或易于污染的产品进行控制的形成文件的安排。
6.风险管理(7.1)。
组织应在产品实现全过程中,建立风险管理的形成文件的要求。应保持风险管理引起的记录。
7.产品要求(7.2.2)。
产品要求得到规定并形成文件。
8.设计和开发程序(7.3.1)。
设计开发策划的输出应形成文件。
9.采购程序(7.4.1)。
10.生产和服务提供的控制。
①必要时,获得形成文件的程序、形成文件的要求、作业书以及引用资料和引用的测量程序(7.5.1.1b)。
②产品的清洁和污染控制的形成文件的要求(7.5.1.2.1)。
③器械安装和安装验接收准则的形成文件的要求(7.5.1.2.2)。
④服务提供活动及其验形成文件的程序、作业书、参考材料和测量程序 (7.5.1.2.3)。
11.计算机软件确认程序及过程确认程序(7.5.2.1)。
12.产品标识程序(7.5.3.1)。
13.可追溯性程序(7.5.3.2.1)。
14.产品防护程序或作业书(7.5.5)。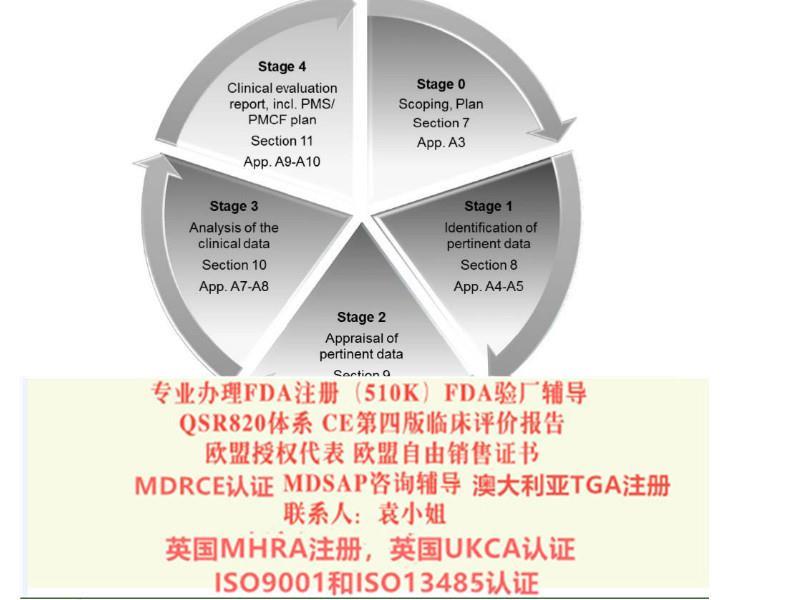
ISO9001和ISO13485的咨询流程
(一)调研诊断。根据接受咨询服务组织的规模、产品和服务复杂程度确定时间,一般控制在三个工作日左右。
(二)分析策划,由咨询师与接受咨询服务组织协商完成。
(三)培训。至少应进行如下培训:
1.标准培训:对接受咨询服务组织、骨干进行标准及管理体系知识普及培训。
2.内审员培训:培训内容要包括标准内容和内审知识,对内审员进行考试,试卷及结果应进行存档,作为内审员培训书的发依据。
(四)管理体系的分析与评审。咨询人员应当与接受咨询服务组织共同分析和评审管理体系的现状,对照标准要求确定改进和完善管理体系的方向。
(五)体系文件编写。咨询人员提出拟编写的文件清单,提出文件编写的要求和时限。接受咨询服务组织文件编写人员起草管理体系文件,咨询组采取适当方式文件编写过程。体系文件既要符合所选择的管理体系标准要求,又要符合接受咨询服务组织的实际情况,同时符合相关法律法规和行政管理要求。
(六)接受咨询服务组织发布体系文件,确定文件发布形式、参加人员、时间安排等。
(七)体系运行。管理体系试运行时间必须达到三个月以上,咨询组现场文件执行、生产/服务现场管理、质量记录使用、保存、归档等工作。体系运行前,咨询师应要求接受咨询服务组织策划并开展体系文件学习培训。
(八)内部审核。由管理者代表与相关部门策划审核方案,编制年度内审计划,确定内审组长,由内审组长编制内审实施计划,并组织内审员编制检查表。咨询人员内审组实施内审,完成相关工作。
(九)管理评审。在次内审后,由接受咨询服务组织立完成管理评审,咨询组以适当形式进行。
(十)模拟审核/*二次内审。模拟审核由咨询机构选派模拟审核组与咨询组、接受咨询服务组织共同完成;模拟审核的相关记录由模拟审核组长负责汇总,提交到技术会审核后交咨询机构存档、备查;认咨询机构可视具体情况,用*二次内审来代替模拟审核。
(十一)认审核的准备。
(十二)认审核不合格项的整改。
(十三)根据合同为接受咨询服务组织提供后续服务。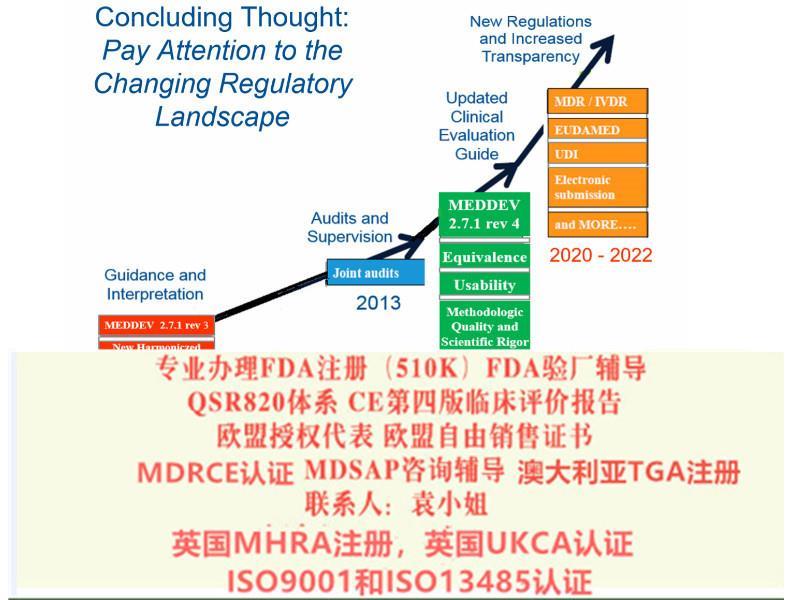
我公司办理产品出口欧盟、美国、中东南美等国家的各种认:
TUV莱茵,TUV南德,***等CE认,CE技术文件编订, CE*四版评价(MEDDEV 2.7.1 Rev 4)编写,欧盟授权代表,欧盟自由销售书,ISO13485:2016,器械单一审核程序(MDSAP)审核、美国FDA注册(含FDA510K申请), FDA QSR820验厂及整改,FDA警告信应对&RED LIST REMOVAL/QSR820体系/OTC验厂及整改,英国BRC认咨询,BSCI验厂;口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试、防护服type5/6 (PPE认咨询(89/686/EC个人防护指令)。
ISO 13485:2016 Medical devices——Quality management systems——Requirements for regulatory purposes(器械 质量管理体系 用于法规的要求),是2019年11月为止的现行版本。
----以下为旧版本介绍,已作废。部分内容仍可参考。-----
ISO13485:2003标准的全称是《器械 质量管理体系 用于法规的要求》(Medical device-Quality management system-requirements for regulatory)。该标准由SCA/TC221器械质量管理和通用要求标准化技术会制定,是以ISO9001:2000为基础的立标准。标准规定了对相关组织的质量管理体系要求,但并不是ISO9001标准在器械行业中的实施指南。
该标准自1996年发布以来,得到全世界广泛的实施和应用,新版ISO13485标准于2003年7月3日正式发布。与ISO9001:2000标准不同,ISO13485:2003是适用于法规环境下的管理标准:从名称上即明确是用于法规的质量管理体系要求。器械在国际上不仅只是一般的上市商品在商业环境中运行,它还要受到国家和地区法律、法规的监督管理,如美国的FDA、欧盟的MDD(欧盟器械指令)、中国的《器械监督管理条例》。因此,该标准必须受法律约束,在法规环境下运行,同时必须充分考虑器械产品的风险,要求在器械产品实现全过程中进行风险管理。所以除了要求外,可以说ISO13485实际上是器械法规环境下的ISO9001。
美国、加拿大和欧洲普遍以ISO 9001,、EN 46001或 ISO 13485作为质量保体系的要求,建立器械质量保体系均以这些标准为基础。器械要进入北美,欧洲或不同国家的市场,应遵守相应的法规要求。
我公司办理产品出口欧盟、美国、中东南美等国家的各种认:
TUV莱茵,TUV南德,***等CE认,CE技术文件编订, CE*四版评价(MEDDEV 2.7.1 Rev 4)编写,欧盟授权代表,欧盟自由销售书,ISO13485:2016,器械单一审核程序(MDSAP)审核、美国FDA注册(含FDA510K申请), FDA QSR820验厂及整改,FDA警告信应对&RED LIST REMOVAL/QSR820体系/OTC验厂及整改,英国BRC认咨询,BSCI验厂;口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试、防护服type5/6 (PPE认咨询(89/686/EC个人防护指令)。
一、新版标准有哪些变化?
ISO13485:2016生命周期的主题已经引入,贯穿产品设计开发,生产,储存,安装和处置的每个阶段,风险管理得到加强,许多要求已被纳入。
以下是ISO13485:2016的主要修订和变化,可作为企业进行ISO13485:2016转换的参考:
1、范围清楚地解释到其适用于产品的存储和分配,内部和外部供应商服务,以及相关服务,例如:设备分销商,运输服务,灭菌服务,安装维护服务,软硬件配件供应商。
2删减条款更为合理。
新版延伸了不适用的条款,允许客户在有合理原因下删减*6,7或8条。这种变化符合当前的业务发展模式,反映了标准的适用性。
3、术语和定义更贴近实际。
条款已经被添加及修改。2003版的定义已经被修改和。新版本包括评估,生命周期,制造商,进口商,分销商,授权代表,绩效评估,投诉,上市后的,风险和风险管理等新技术术语。
4、条款变更的更为合理。
新版本更多地考虑到的要求,并强调需遵守要求,以确保设备的*性和有效性。
许多要求已被采纳,如美国的FDAQSR820,日本的JPAL MO 69,巴西GMP,EU-MDR和EU-IVDR以及加拿大,澳大利亚和中国的要求,使其具有更广泛适用性。
例如,介绍了可用性和软件应用要求; 控制设计过程更为详细; 明确规定了变更控制的要求; 加强对供应商管理的要求; 定义了可追溯性(UDI)的要求和目的; 添加了反馈和投诉处理的要求。
5.、风险管理的要求得到加强,新版本更加明确。
许多地方都提到“风险”和“风险管理”。提出对供应商风险的控制。器械风险,反馈机制,投诉处理和数据分析更为详细,从而使得上市后的风险系统更加可操作。
http://sungofda.cn.b2b168.com
欢迎来到上海沙格企业管理咨询有限公司网站, 具体地址是上海市金山区石化街道松南支路48号-826座,联系人是袁小姐。
主要经营上海沙格企业管理有限公司SUNGO是欧盟授权代表,从事FDA验厂、欧盟自由销售书、医疗器械单一体系审核MDSAP、CE*四版临床评价报告、MDRCE认、CE MDR认、CE技术文件、EU 2017/745认、MDEL注册、MDR、CE认等服务。。
单位注册资金单位注册资金人民币 100 万元以下。
我们公司主要提供CE,MDR认,MDR,CE认,IVDR,欧代,EU2017/745认,2017/746,EC,REP,CE技术文件,CE*四版临床评估报告.SUNGO,EN,455,UK,EN12184,清洗验,FDA验厂等服务,我们确信,凭借我们的专业服务和良好的协调、沟通能力,使客户在经营生产中顺利进行,协助客户不断成长,在合作中与客户实现共赢。欢迎您致电咨询!