产地上海
可售地**
品牌SUNGO
型号SUNGO BV
包装纸质
级医用防护服、隔离衣、手术衣, 完成公告机构现场审核及审核中不符合项的整改;
法规条款增加,认证评审更加严格
a. 分类规则增加:由MDD的18条增加到 22条;
b. 基本要求检查表条目增加:由MDD的13条增加到 MDR的23条;
c. CE技术文件的结构发生了变化,分为:产品技术文件和上市后文件(MDD只要求产品技术文件);
d.评价报告。MDR要求企业提供第四版评估报告,相比于第三版,第四版要求更为严格;
5:MDR要求更高的透明度和可追溯性
a. 引入了器械标识UDI,增加产品的可追溯性;
b. 企业的相关信息都会被收集到欧洲器械数据库(EUDAMED);
c. 建立上市后监督(PMS)系统;

如果假设发现潜在的化学变化和观察到的物理性能变化之间有直接的关系,那么公式(B.3)也可作为物理性能达到规定阈值所需时间的模型。
如果阿列纽斯公式适用,那么公式(B.3)可得到由对l/T(K-1)组成的坐标系上的一条直线。假设得到了这条直线,那么很*外推这条线,并确定目标温度下发生预先确定程度的变化所需时间。活化能EA可由识别的线的斜率*地计算出来:

关于Free sale certificate自由销售证书,器械办理自由销售证书有三种:
种:中国食品药品监督管理局出局的器械产品出口销售
(这种办理条件:企业必须是生产企业,有国内的产品注册证&备案证,生产企业许可证&备案证,齐全,企业直接联系局,递交资料可以申请,不需要费用)
第二种:也是需要有注册证和许可证,可以由品商会这些出局的自由销售证书
(这种一般泰国,越南这些,也认可商会这种自由销售证书)
周期是一个半月左右
第三种:对于企业是否有中国的器械注册证和生产许可证,并没有特别的要求的。
我们市场常见 英国,德国 美国 荷兰等等这些发达出的自由销售证书的,这种尤其是被沙特,埃及,阿根廷等
这种主要看产品,如果在欧盟风险等级属于I类的话,不需要国内局的任何,配合填写申请表,说明书,符合性声明即可
如果在欧盟的风险等级属于II类的话,不需要国内局的任何,但是必须要有公告机构的CE证书才可以办理。
十二、完善评价相关要求
新法规提出:
要求根据Article61和附录XIV partA执行、评估、报告和更新评价资料;
提出对特定III类和IIb类器械,CER中要考虑咨询小组的意见;
对植入和III类器械,提出考虑研究;
要求CER按照PMCF取得数据进行更新;
针对III类和可植入器械,提出了CER更新的频率;
明确实质等同性需考虑的特点;
要求其与风险管理的相互作用
十三、Eudamed数据库
新法规提出:
明确欧洲器械数据库(Eudamed)建立目的和包含的信息(Article 33);
信息的公开性:
要求III类器械和植入式器械,和性能信息通过Eudamed向公众开放。
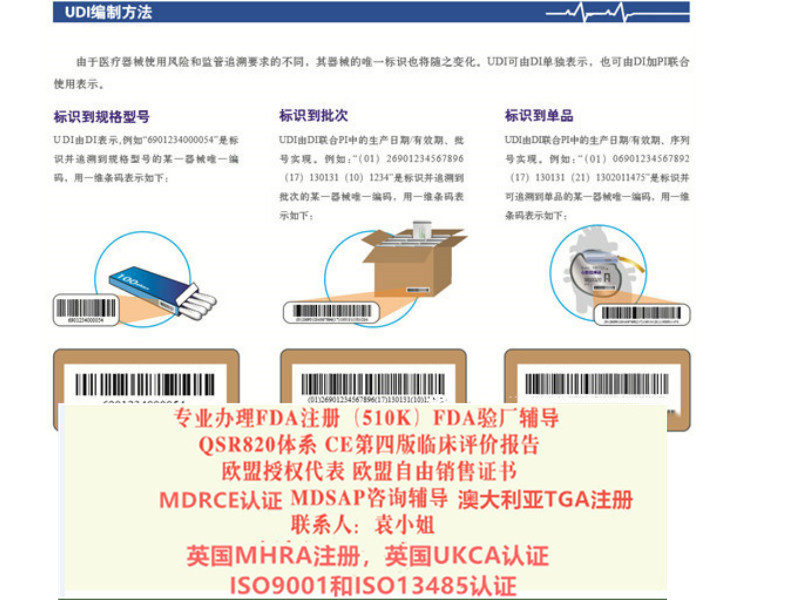
十四、提出器械的可追溯性(UDI)
除定制和研究器械外,其他器械均需建立UDI系统;
UDI信息体现在标签或包装上(不包含集装箱);
UDI-DI信息需要载明于符合性声明中(见Article27);
Annex VI Part B提出UDI-DI包含的信息;
可植入、重复使用、软件、可配置器械的UDI有要求(见Annex VI Part C)
包装或标签上UDI实施的时间见Article123 (f)。
UDI 发行实体由欧盟会。
过渡性:Article 120指出“在会根据第27(2)条发行实体前,GS1、HIBCC和ICCBBA应被视为的发行实体”。
加强警戒和市场,一旦器械可以在市场上使用,制造商将必须收集有关其性能的数据,欧盟国家将在市场领域进行更密切的协调。
http://sungofda.cn.b2b168.com










