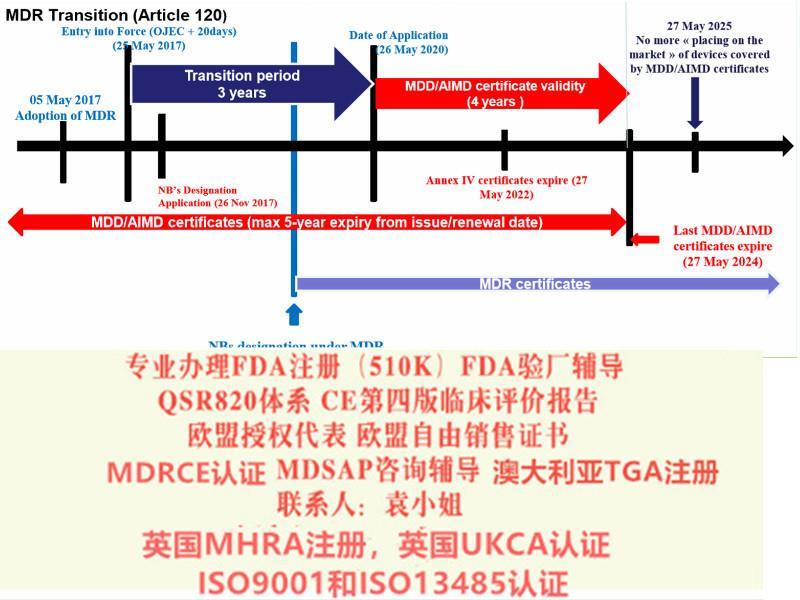
2024年5月25日:AIMD,MDD和IVDD书将失效

MDR法规要求CE认NB必须要有自己的审核*团队,新法规下对外聘*的做法将有所限制;
MDR将有源器械指令纳入了进来,与一般器械指令(93/42/EEC)合二为一
医疗器械MDRCE认和MDD指令CE认区别

MDR纳入更多产品进行管理;MDR分类规则的主要变化
MDR新法规生效后, NB将按照新的资质要求重新授权,不符合要求的NB将会被淘汰。

MDR法规要求申请CE认NB必须要有自己的临床*,而不能仅靠外部临床*进行相关审核。

出口:美国FDA注册(含FDA510K申请)、 FDA QSR820验厂及整改、FDA警告信应对&RED LIST REMOVAL/QSR820体系/OTC验厂及整改、CE认(CE整套技术文件编订、 CE第四版临床评价(MEDDEV 2.7.1 Rev 4)编写)、ISO13485:2016、欧盟授权代表、欧盟销售书、英国BRC认,BSCI验厂;口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试、防护服type5/6 (PPE认(89/686/EC个人防护指令)。
制造商的法律责任
制造商是指以他自己名义设计、生产、翻新、销售器械的个人或法人。
- 产品标签、说明书、包装上的制造商要承担相应的法律责任,且产品本身一定是在自己工厂生产的;
- 必须确保产品的设计和生产过程符合MDR&IVDR的要求;
- 必须建立、文件化、落实、保持风险管理的要求;
- 必须按规定对产品进行临床/性能评估,包括上市后临床跟踪/性能跟踪;
- 必须按MDR&IVDR附录II+III建立CE技术文件
- 应签署符合性声明,正确使用CE标志;
- 应使用UDI系统及履行注册责任;
- 应将CE技术文件、CE书、符合性声明保存到后一个产品出厂后5-10年,III类器械保存15年,并以供主管当局的检查;
- 建立上市后监督系统,不断更新信息;不断改进质量管理体系,确保标准及技术规范变化后,能及时改进产品;
- 应确保器械标签、说明书使用欧盟成员国*的语言;
- 当产品不符合MDR&IVDR要求时,制造商应及时采取纠正措施,对产重问题应及时通知主管当局和公告机构;
- 应建立事故报告和FSCA的制度;
- 主管当局提出时,制造商有义务以欧盟语言提品符合要求的资料,需要时,免费提供样品;
- 如果产品是委托其他人设计和生产的,应当将相关信息提供给欧盟数据库;
- 如果产品适成人员伤害了,制造商应承担赔偿责任,因此要做好财务上的安排;
- 至少任命一名负责法规合规性的员工,应有4年以上器械法规事务或质量管理体系工作经验,或理工科、学、医学大学以上*加1年以上的相关工作经验
MDR法规对NB公告机构的监管要求(新法规生效后NB将按照新的资质要求重新进行授权)

MDRCE认:CE整套技术文件编订、 CE第四版临床评价MEDDEV 2.7.1 Rev 4)
-/gjhdff/-
http://sungofda.cn.b2b168.com