1. FDA注册的日常维持,更新;
2. FDA较新动态的**时间告知及专业建议;
3. 随时为您提供与FDA注册和上市后监管等相关的事务的处理意见;
4.可代表您回答您美国客户的相关FDA问题(英文邮件,英文电话会议均可);
5.接受FDA验厂时,以优惠的价格提供QSR820辅导、审核经验传授及全程英语陪审
6.选择有能力进行QSR820辅导(包括美国法规知识、国内审核经验和英语沟通能力)的美国代理人!
7.在完成注册后,启动QSR820体系,按照美国法规的要求实施管理,有备无患!
8.实在没有满足第2条又被抽查到,立即联系我们,为你提供五天辅导方案确*审查!
专业提供医疗FDA注册,食品FDA注册、化妆品FDA注册,美国代理人服务、FDA验厂辅导、FDA警告性移除
FDA官方对于美国代理人的解释也可以参考看一下的原文:
Any foreign establishment engaged in the manufacture, preparation, propagation, compounding, or processing of a device imported into the United States must identify a United States agent (U.S. agent) for that establishment.
Information about a foreign establishment’s U.S. Agent is submitted electronically using the FDA Unified Registration and Listing System (FURLS system) and is part of the establishment registration process. Each foreign establishment may designate only one U.S. agent. The foreign establishment may also, but is not required to, designate its U.S. agent as its official correspondent. The foreign establishment should provide the name, address, telephone and fax numbers, and e-mail address of the U.S. agent.
The U.S. agent identified will be required to complete an automated process to confirm that they have agreed to act as the U.S. agent. The automated process will forward an email verification request to the U.S. agent. They will be requested to confirm her/his consent to act as a representative/liaison on behalf of the foreign establishment. If the U.S. agent denies consent (or does not respond within 10 business days), the Official Correspondent/Owner Operator of the foreign establishment will be notified and must designate a new U.S. agent to satisfy the regulatory obligation.
Responsibilities of a U.S. agent
The U.S. agent must either reside in the U.S. or maintain a place of business in the U.S. The U.S. agent cannot use a post office box as an address. The U.S. agent cannot use just an answering service. They must be available to answer the phone or have an employee available to answer the phone during normal business hours.
The responsibilities of the U.S. agent are limited and include:
assisting FDA in communications with the foreign establishment,
responding to questions concerning the foreign establishment's devices that are imported or offered for import into the United States,
assisting FDA in scheduling inspections of the foreign establishment and
if FDA is unable to contact the foreign establishment directly or expeditiously, FDA may provide information or documents to the U.S. agent, and such an action shall be considered to be equivalent to providing the same information or documents to the foreign establishment.
Please note that the U.S. agent has no responsibility related to reporting of adverse events under the Medical Device Reporting regulation (21 CFR Part 803), or submitting 510(k) Premarket Notifications (21 CFR Part 807, Subpart E).
我公司专业办理医疗产品出口欧盟、美国、中东南美等国家的各种认证:
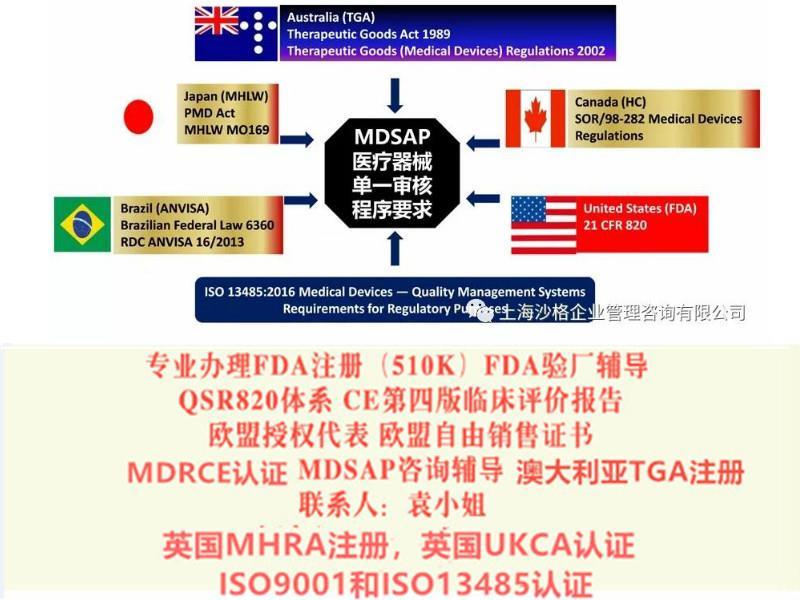
TUV莱茵,TUV南德,***等CE认证(MDD/MDR法规),全套CE技术文件编订, CE第四版临床评价(MEDDEV 2.7.1 Rev 4)编写,欧盟授权代表,欧盟自由销售证书,ISO13485:二016,医疗器械单一审核程序(MDSAP)审核、美国FDA注册(含FDA510K申请),FDA美国代理人服务/ FDA QSR820验厂辅导及整改,FDA警告信应对&RED LIST REMOVAL/QSR820体系辅导/OTC验厂辅导及整改,英国BRC认证咨询,BSCI验厂辅导;口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试、防护服type5/6 (PPE认证咨询(89/686/EC个人防护指令辅导)。
医疗器械美国FDA 510(k)认证简介
为了在美国上市医疗器械,制造商必须经过两个评估过程其中之一:上市前通知书[510(k)](如果没有被510(k)赦免),或者上市前批准(PMA)。 大多数在美国进行商业分销的医疗器械都是通过上市前通知书[510(k)]的形式得到批准的。在某些情况下,在1976年5月28日之前合法上市的器械,既不要求递交510(k)也不要求递交PMA。
510(k)文件是向FDA递交的上市前申请文件,目的是证明申请上市的器械与不受上市前批准(PMA)影响的合法上市器械同样*有效,即为等价器械(substantially equivalent)。申请者必须把申请上市的器械与现在美国市场上一种或多种相似器械对比,得出并且支持等价器械的结论。合法上市器械是在1976年5月28日之前合法上市的器械(preamendment device),或者从III类器械中分入II或I类的器械,或者通过510(k)程序发现与这样的器械等价的器械,或者通过自动的III 类器械定义的评价建立的器械。与之等价的器械被称为“predicate device(s)”。 申请者必须提交描述性的数据,必要的时候,要提交性能数据来说明器械是predicate device的等价器械。再次说明,510(k)的数据是显示相似性的数据,即,新器械与predicate device的等价程度。
FDA 等价器械
510(k)不像PMA那样要求合理的*性和有效性的证明,而是要求等价器械的证明。等价器械就是新的器械与predicate device一样*有效。与predicate device相比,如果符合下列条件,就认为器械是等价器械:
与predicate device有相同的使用目的,具有相同的技术性能;或者
与predicate device有相同的使用目的,具有不同的技术性能,但是并没有增加*性和有效性的问题,并且证明人证明器械与合法上市器械一样*有效。
所谓等价器械并不是说新的器械与predicate devices必须完全相同。等价器械是关于使用目的、设计、使用的或传送的能源、材料、性能、*性、有效性、标注、生物相容性、标准和其他可应用的特征。
申请者在收到宣布为等价器械的指令之前,器械不得上市。一旦器械确定为等价器械,然后就可以在美国上市。如果FDA确定器械不是等价器械,申请者可以递交另一份含有新数据的510(k)文件,提出重新分类请求,或者递交上市前批准申请(PMA)。通常在90天内,基于申请者递交的信息,得出等价器械的结论。
谁必须递交510(k)
食品、药品和化妆品(FD&C)行动**和21 CFR 807的510(k)规章中并没有特别指出谁必须申请510(k)——任何人都可以申请。但是,他们*了哪种行为,例如把器械引入美国市场,要求510(k)申请。
基于*的行为,必须向FDA递交510(k)的如下所示:
1) 把器械引入美国市场的国内厂家;
如果成品器械厂家根据他们自己的规范装配器械,并在美国上市,那么必须递交510(k)。然而,器械组件厂家并不要求递交510(k),除非这些组件销售给终用户作为替换零件。合同厂家,这些公司根据其他的规范按照合同装配器械,不要求递交510(k)。
2) 把器械引入美国市场的规范制订者;
FDA审查规范制订者与审查厂家几乎一样。规范制订者是制订成品器械规范的人,但是器械按照合同由其他的公司来生产。因此,规范的制订者,而不是合同厂家需要递交510(k)。
3) 改变标注或操作严重影响器械的再包装者或再标注者;
如果再包装者或再标注者严重改变了标注或影响了器械的其他条件,可能会要求递交上市前通知书。此时,你必须确定是否通过修改指南,删除或增加了警告,禁忌征候等等而显著改变了标注,还有包装操作是否能够改变器械的条件。然而,大多数的再包装者或再标注者并不要求递交510(k)。
4) 把器械引入美国市场的外国厂家/出口商或外国厂家/出口商的美国代理方。
何时需要510(k)
在下列情况下需要递交510(k):
**次进行商业分配(上市)。在1976年5月28日之后(FD&C Act进行医疗器械修正的有效日期),任何想在美国出售医疗器械的人都要求在器械上市之前至少90天递交510(k)申请。如果器械在1976年5月28日之前不是由你的公司上市的,要求递交510(k)。
对于已上市器械提出不同的使用目的。510(k)规范(21 CFR 807)对于使用目的的主要变化,特别要求递交上市前通知书。使用目的在器械的标注或广告的声明中指出。然而,如果使用意图没有全部发生变化,大多数的变化都需要递交510(k)。
已上市器械发生改变或改进,如果这个改变能够严重影响器械的*性或有效性的情况下。
申请人负责决定改进是否能够严重影响器械的*性或有效性。无论得出怎样的结论,都要做纪录,此记录能够在器械主记录中反应出来,在医疗器械质量管理规范的要求下,改变控制记录。如果被询问到,申请者就能够证明评估了这个改动。
对现有器械进行了显著影响器械*性或有效性的改变或改动,或者上市器械的指南为全新的,或与原来不同的情况下,要求递交新的,完整的510(k)文件。
何时*510(k)
下面情况下* 510(k):
如果器械厂家向另一个企业卖没有完工的器械,要求进一步加工,其中包括用于其它企业组装器械的零件的情况下,不需要递交510(k)。 然而,如果生产的零件是要直接卖给终端用户作为替代零件,就需要510(k)。
如果生产的器械不上市或不进行商业分发,就不需要 510(k)评估或检验器械。这包括临床评估。如果生产的器械用于进行临床试验,则可能受到研究用器械赦免(IDE)法规的管理。
如果分销其他公司国内生产的器械,代理商不需要递交510(k)。代理商可以把“Distributed by ABC Firm”的标签贴在器械上,卖给终端用户而不用递交510(k)。
大多数情况下,如果器械现有的标签或条件没有显著改变,那么再包装者或再标注者就不要求递交510(k)。
如果器械是在1976年5月28日之前合法上市的,就不用递交510(k)文件,除非进行了改进或使用目的上有变化。这些器械被称为“grandfathered”。
如果是外国制造器械的进口商,在下列情况下不需要递交510(k):
510(k)已经由外国厂家递交,并得到上市批准,或
510(k) 已经由进口商代表外国厂商递交了,并得到上市批准。如果一个进口商代表外国厂商递交了510(k),那么所有从相同的国外厂商(510(k)持有人)进口相同器械的其他进口商就不要求递交此器械的510(k)文件。
某些Ⅰ类或Ⅱ类器械在**次上市时可以不递交510(k)。Ⅰ类和Ⅱ类赦免器械的规范可以在医疗器械赦免中找到。
我公司专业办理医疗产品出口欧盟、美国、中东南美等国家的各种认证:
TUV莱茵,TUV南德,***等CE认证(MDD/MDR法规),全套CE技术文件编订, CE第四版临床评价(MEDDEV 2.7.1 Rev 4)编写,欧盟授权代表,欧盟自由销售证书,ISO13485:二016,医疗器械单一审核程序(MDSAP)审核、美国FDA注册(含FDA510K申请),FDA美国代理人服务/ FDA QSR820验厂辅导及整改,FDA警告信应对&RED LIST REMOVAL/QSR820体系辅导/OTC验厂辅导及整改,英国BRC认证咨询,BSCI验厂辅导;口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试、防护服type5/6 (PPE认证咨询(89/686/EC个人防护指令辅导)。

1:FDA 化妆品验厂应对
依据FDA指南文件Guidance forIndustry Cosmetic Good Manufacturing PracticesFOOD AND DRUG ADMINISTRATIONCOMPLIANCE PROGRAMGUIDANCEMANUAL -COSMETIC MANUFACTURING INSPECTIONS
2:食品企业出口到美国在进行了企业注册之后,可能会被美国FDA抽查到验厂。 验厂会由美国评审员进行,评审的重点是现场的GMP和SSOP以及HACCP计划,同时批记录也是关注的重点。如果审核失败,或者你不接受审核都将导致你失去美国市场。我公司有数家成功验厂辅导经验可以协助企业准备应对FDA的验厂我公司可以提供FDA 验厂辅导、审核陪同和翻译服务:
3:医疗行业 收到FDA当局发来的验厂通知怎么办?
FDA验厂辅导(QSR820)QSR820,又称21CFR820,是美国医疗器械质量管理体系法规的英文缩写,因其位于美国联邦法规(Codeof Federal Regulations)第21卷第820部分,故名。QSR820是美国(人用)医疗器械制造商以及拟将产品销往美国的外国(人用)医疗器械制造商必须遵守的质量管理体系法规。是多数医疗器械在美国上市之前必须遵守、上市之后随时可能抽查的基本要求。这种抽查即通常所说的FDA工厂检查(医疗器械)。
FDA当局在北京成立办事处之后,表明将会不定期的对FDA企业进行抽查,即使审核通知从之前的提前2个月变为提前5个工作日通知,我司也能帮助企业积极应对FDA的审核检查,我司厦门一家客户被北京FDA办事处突击抽查,紧张的5个工作日准备之后迎接来了FDA的审核,较终以零不符合项通过检查,实力足以见证详细需要的话
4:药品企业收到FDA当局发来的验厂通知怎么办专业应对FDA抽查审厂审核、改善、审核陪同,
FDA验厂依据OTC 质量体系指南符合性指导OTC 质量体系指南符合性指导:
我们提供的服务:
现已有质量管理体系与法规要求的差距
咨询过程的整体设计,包括现场的整改以及文件化体系以及应用的整改;
收集现有的文件资料:质量手册,程序文件,作业指导书,工艺文件,检验规程,记录等
基于FDA审核要求文件记录审阅、检查生产全过程GMP、设备设施维持GMP内容,咨询师与企业相关人员一起进行文件系统修整;
帮助企业发现车间和仓库的不足与整改;
对企业人员进行迎接审核技巧的培训;
体系有效性的检查,在FDA来审核之前,安排本公司评审员进行模拟审核;
陪同FDA验厂;
协助企业进行不符合项的整改
我公司专业办理医疗产品出口欧盟、美国、中东南美等国家的各种认证:
TUV莱茵,TUV南德,***等CE认证(MDD/MDR法规),全套CE技术文件编订, CE第四版临床评价(MEDDEV 2.7.1 Rev 4)编写,欧盟授权代表,欧盟自由销售证书,ISO13485:二016,医疗器械单一审核程序(MDSAP)审核、美国FDA注册(含FDA510K申请),FDA美国代理人服务/ FDA QSR820验厂辅导及整改,FDA警告信应对&RED LIST REMOVAL/QSR820体系辅导/OTC验厂辅导及整改,英国BRC认证咨询,BSCI验厂辅导;口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试、防护服type5/6 (PPE认证咨询(89/686/EC个人防护指令辅导)。
我公司专业办理 FDA注册,FDA验厂辅导、医疗器械FDA QSR820验厂辅导、OTC CGMP验厂辅导及食品FDA验厂辅导 、培训、翻译,FDA警告信处理,FDA黑名单移除
我公司有丰富的医疗器械FDA QSR820验厂辅导、OTC CGMP验厂辅导及食品FDA验厂辅导经验,咨询师资源既具备深厚的法规背景知识,又具备非常强的专业英文能力,与FDA的沟通非常顺畅,对于FDA的审核思路和开具不符合的意思理解深刻,整改到位。

-/gjhdff/-
http://sungofda.cn.b2b168.com