产地上海
可售地**
品牌SUNGO
型号SUNGO BV
包装纸质
欧代和注册服务SUNGO 荷兰和德国公司可以提供欧盟授权代表服务,同时提供向当地部门申报注册的服务。
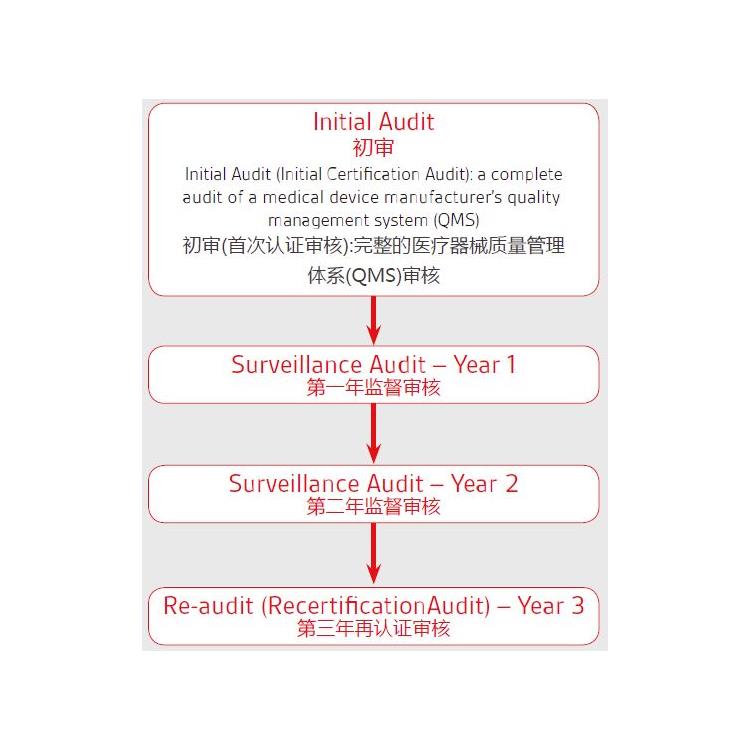
按产品的危险程度,将产品分为Ⅰ类、Is(),Im(测量),Ⅱa类、Ⅱb类、Ⅲ类
对于目前MDD和MDR的各个风险等级的产品实施时间:
1. 对于I类的产品,欧盟强制要求2021年5月25号强制实施MDR
2. 对于Is(),Im(测量),Ⅱa类、Ⅱb类、Ⅲ类迟可以用到2024年5月25号
3. 对于可重复使用的产品,在MDD里属于I类,但是在MDR里属于Ir类,那么如果企业在2021年5月25号之前满足了MDD的要求,迟可以用到2024年5月25号
上市后(PMS) (MDR第83~86条)
PMS需收集、记录并分析器械在其整个生命周期内的质量、性能和相关数据,以得出必要的结论,并确定、实施和监测任何预防及纠正措施。
I类器械制造商应编制一份上市后报告,总结根据上市后计划收集的数据分析结果和结论,以及采取的任何预防和纠正措施的理由和说明。必要时应更新报告,并应主管机构的要求提供。
IIa、IIb和III类器械制造商应针对各器械或类别或器械组编制定期性更新报告(PSUR),总结根据上市后计划收集的数据分析结果和结论,并对采取的任何预防和纠正措施提供理由和说明。

科学的理念体现在法规的细节规定或管理要求中。如器械产品种类繁多,法规对于类别产品,其符合性评估程序中分别规定了要求,例如与一同使用的器械的认程序、利用人类或动物源组织或细胞及其物制造器械时的认程序等,体现产品的个性化要求;对于高风险产品,欧盟法规规定了小组的职责中包含前咨询的程序;

MDD指令和MDR法规的CE认的区别
1:老MDD指令申请CE认,由于法规规定产品在市场上出现任何问题,都是由制造商承担。其中欧盟授权代表的职责只是沟通协调以及产品包装可以使用欧盟授权代表的公司名字和地址信息的责任。国外的进口商更多的是找工厂要一张MDD的CE书,能顺利清关销售便可以了,一般不关注你们这个书怎么获得的,是否正真满足法规要求的。
2:但是新MDR的管控趋于严格,对于制造商,欧盟授权代表以及国外进口商三方该承担的责任比较明确,欧盟授权代表和进口商与制造商一样为缺陷器械承担连带的法律责任。所以进口商在采购工厂产品的时候,较MDD老法规,他们更关注,工厂是否真正满足CE法规要求,尽量的将自己要承担的风险降低到。
3:我们为企业编写的MDR CE技术文件里的:风险分析报告,评价报告,基本基本检查表等等,不仅仅是为了获得一张书而做的,更多的都是从各个方面来产品是的有效的。
需要办理以下认可以随时找我 :
1:出口欧盟:MDR CE认/IVDRCE认,欧盟授权代表,欧盟注册,欧盟自由销售书
2:出口英国:英国代表,英国MHRA注册,UKCA认,英国自由销售书
3:出口美国:美国FDA注册,FDA510K,QSR820体系
4:中国:国内的器械注册和生产许可
5:出口加拿大:加拿大的MDEL注册
6:质量管理体系认:ISO13485咨询和认
我公司办理:出口瑞士:需要瑞士代表,瑞士注册
http://sungofda.cn.b2b168.com