创可贴EC REP要求 SUNGO的瑞士代表注册 英国代表是什么
更新时间:2024-05-13 浏览数:45
所属行业:
咨询 管理咨询
发货地址:上海市金山区
产品数量:9999.00个
价格:面议
周期4周
包装纸质
品牌SUNGO
是否进口否
可售卖地**
SUNGO是**化的器械法规技术服务商。从事行业服务达13年,累计服务客户**过4000家。在美国FDA注册、欧洲注册以及中低风险器械的认证业务上,市场份额长期稳居行业位,得到客户普遍**。选择SUNGO,不是选择了一次性的合作伙伴,而是选择了一个长期的技术支持的战略伙伴。
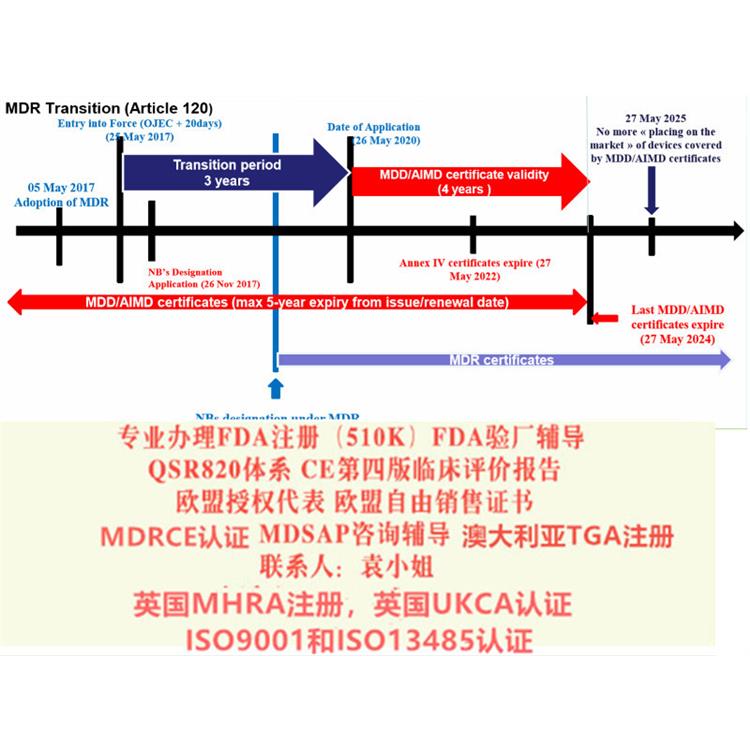
MDR的主要变化 1.扩大了应用范围 2.提出了新的概念和器械的定义 3.细化了器械的分类 4.完善了器械的通用和性能要求 5.加强对技术文件的要求 6.加强器械上市后的 7.完善评价相关要求 8.提出Eudamed数据库的建立和使用 9.提出器械的可追溯性(UDI) 10.对NB提出严格的要求在新版MDR 2017/745/EU中,更是完善了评估(包括器械售后追踪)和调查的执行、评估、报告和更新资料的相关要求。对特定III类和IIb类器械,评估报告中要考虑咨询小组的意见;对植入式和III类器械,提出考虑研究;要求评估报告按照售后追踪所取得的数据进行更新;

主要变化之九:风险-收益 附件7提供了详细的指南,对于器械的性和性能表述; 附件7.2 讨论了风险和收益分析,包括对于风险和收益的量的评估,以及总体评价。交付后的数据价值,以及可能会影响统计有效性行的因素等。

上市后(PMS) (MDR第83~86条) PMS需收集、记录并分析器械在其整个生命周期内的质量、性能和相关数据,以得出必要的结论,并确定、实施和监测任何预防及纠正措施。 I类器械制造商应编制一份上市后报告,总结根据上市后计划收集的数据分析结果和结论,以及采取的任何预防和纠正措施的理由和说明。必要时应更新报告,并应主管机构的要求提供。 IIa、IIb和III类器械制造商应针对各器械或类别或器械组编制定期性更新报告(PSUR),总结根据上市后计划收集的数据分析结果和结论,并对采取的任何预防和纠正措施提供理由和说明。

我们的服务包括: 确定您的器械在加拿大的具体分类。 代表您准备加拿大器械许可证(MDL)或加拿大器械机构许可证(MDEL)申请。 制定、实施或修改您的ISO 13485质量管理体系以满足加拿大的相关规定。 提供关于ISO 13485与加拿大器械法规(CMDR)方面的现场员工培训。 确定每年应向加拿大缴纳的许可证费用。 可以现场审计服务,确保符合ISO 13485:2003以及CMDR相关规定。 了解加拿大器械注册服务的更多信息,请联系我们
级医用防护服、隔离衣、手术衣,按照MDR附录II+附录III的要求编制CE技术文件;
http://sungofda.cn.b2b168.com