产地上海
可售地**
品牌SUNGO
型号SUNGO BV
包装纸质
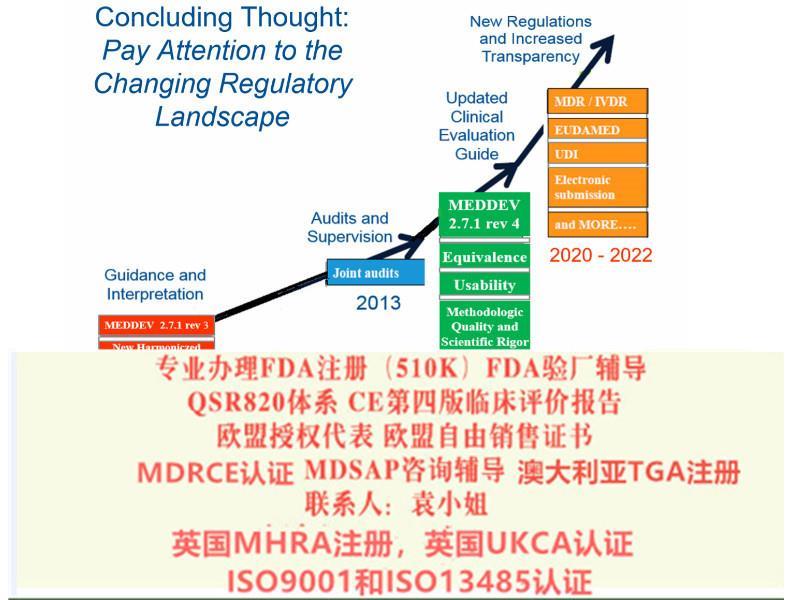
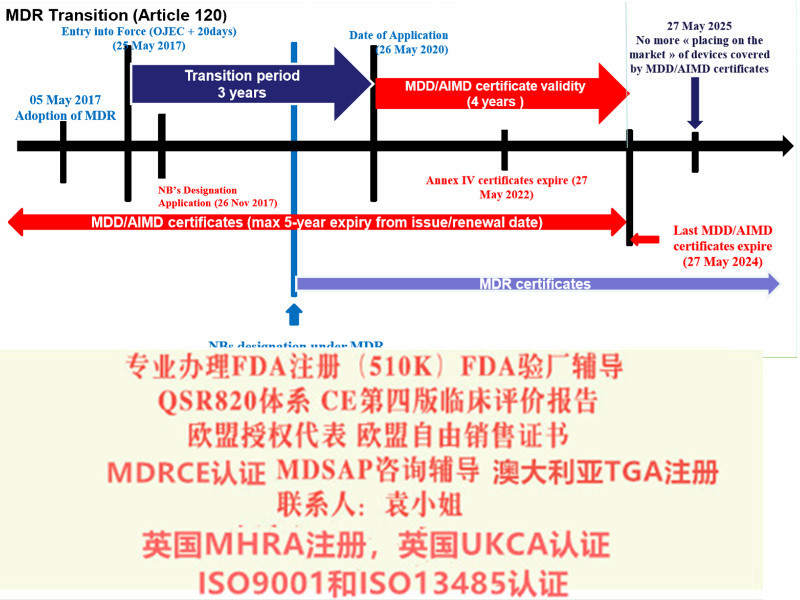
2017年2月Regulation (EU) 2017/745 on Medical Devices器械法规(MDR)提案发布,同年3月,欧盟成员国一致投票表决同意MDR。2017年5月5日,欧盟Official Journal正式对外宣MDR法规内容。MDR新法规将取代现行的有源器械指令Council Directive 90/385/EEC on Active Implantable Medical Devices (AIMDD) (1990)以及器械Council Directive 93/42/EEC on Medical Devices (MDD) (1993)指令。原计划2020年5月26日正式实施的MDR受**影响将推迟实施时间至2021年5月26日。
加拿大MDEL注册
若您计划在加拿大销售器械,您需要进行产品注册登记以获得许可证加拿大颁发两种不同类型的许可证,两种许可分别有不同的要求。
IIa、IIb和III类器械制造商应针对各器械或类别或器械组编制定期性更新报告(PSUR),总结根据上市后计划收集的数据分析结果和结论,并对采取的任何预防和纠正措施提供理由和说明。
IIa类器械制造商应在必要时至少每两年更新PSUR ,IIb和III类器械的制造商应至少每年更新PSUR。
警戒 (MDR第87~92条)

十二、完善评价相关要求
新法规提出:
要求根据Article61和附录XIV partA执行、评估、报告和更新评价资料;
提出对特定III类和IIb类器械,CER中要考虑咨询小组的意见;
对植入和III类器械,提出考虑研究;
要求CER按照PMCF取得数据进行更新;
针对III类和可植入器械,提出了CER更新的频率;
明确实质等同性需考虑的特点;
要求其与风险管理的相互作用
十三、Eudamed数据库
新法规提出:
明确欧洲器械数据库(Eudamed)建立目的和包含的信息(Article 33);
信息的公开性:
要求III类器械和植入式器械,和性能信息通过Eudamed向公众开放。
十四、提出器械的可追溯性(UDI)
除定制和研究器械外,其他器械均需建立UDI系统;
UDI信息体现在标签或包装上(不包含集装箱);
UDI-DI信息需要载明于符合性声明中(见Article27);
Annex VI Part B提出UDI-DI包含的信息;
可植入、重复使用、软件、可配置器械的UDI有要求(见Annex VI Part C)
包装或标签上UDI实施的时间见Article123 (f)。
UDI 发行实体由欧盟会。
过渡性:Article 120指出“在会根据第27(2)条发行实体前,GS1、HIBCC和ICCBBA应被视为的发行实体”。
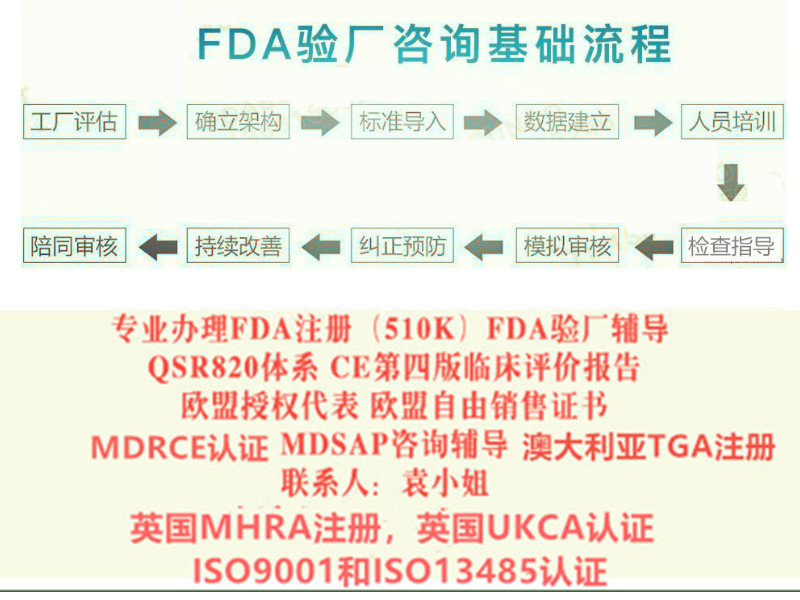
我司是技术顾问+检测 一体的咨询公司,已经成功帮多家申请提供医用口罩的FDA510K技术服务 以及ASTM2100的检测服务
帮企业成功快速的获得FDA510K号
在过渡期结束后,制造商是否仍然可以投放市场/投入使用符合指令的设备? 是的,在某些条件下,可以选择继续投放市场/投入使用符合指令的设备,直到其现有证到期为止。 这可以避免在MDR下立即需要新证。 要使用此选项,所有现有证必须有效(例如,QMS),设备的目的和性质不得更改,并且您必须遵循新的MDR规则进行注册,监视和警惕。
http://sungofda.cn.b2b168.com