周期8周
费用100000
资料基本资料
机构SUNGO
产品医疗用品
欧盟授权代表的名称和地址必须器械随附的信息上,例如(包装)标签和使用说明。如果发生事故,欧盟授权代表将协助并协调向主管当局报告事件。
*各国转换为法律法规即可实施。在内容方面,MDR基于原始指令的整合,大大改进了器械认证的规范和限制,例如产品分类规则,设备可追溯性,性能研究规范,上市后产品性提高以及有效性方面和。 MDR由10章和123篇文章组成,共17个附录。关于的过渡期:仅拥有根据指令90/385/EEC和93/42/EEC颁发的证书的设备可以投放市场,前提是自MDR应用之日起设计和预期用途没有发生重大变化且符合要求新规定。

出口美国需要的为: FDA注册,FDA510K,QSR820体系(美国FDA验厂)

主管当局严格NB对技术文档, 特别是评估的评审1) 强化符合性评审程序, 试验和评估, 警戒系统和市场监督系统, 确保器械的透明和可追溯性2) MDD + AIMD →MDR; IVDD →IVDR3) 药品, 化妆品和食品不在MDR/IVDR的范围内4) MDR包括含有无活性的人组织或细胞的器械5) 无用途, 但功能和风险与器械类似的产品也纳入MDR6) 含纳米材料的器械要进严格的评估定义: Commission Recommendation 2011/696/EU (OJ L 275, 20.10.2011, p.38).7) 电子商务服务 Information Society Service要符合Directive (EU) 2015/15358) 通用规范CS (Common Specification)9) 规定经销链相关方 (Economic Operator) 的职责10) MDD/AIMD中的Annex 的评估 或警戒系统纳入到了MDR的正文里11) 机构内部使用的器械12) 缺陷产品对患者的补偿13) 风险管理和评估互相依赖, 定期更新14) 法规负责人需要满足要求, 负责监督和和控制器械的生产, PMS和警戒系统的活动.
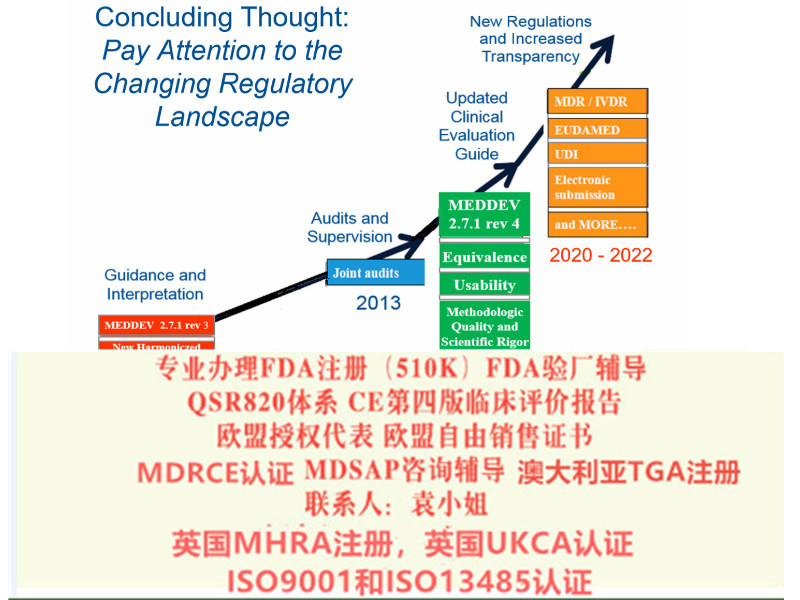
综上所述,持有MDD CE证书的制造商需确认是否有满足过渡条款内提到相关要求,在近一次监督审核前,尽快建立完善相应文件,以保证产品在欧盟市场上的顺畅流通。
加拿大器械机构许可证(MDEL) 假如您生产的是I类器械或IVD,并且不通过经销商而直接销售到加拿大,那么您必须获得器械机构许可证(MDEL)。假如您选择在加拿大通过经销商进行产品销售,那么您的经销商必须拥有MDEL许可证。 无论器械属于哪个类别,器械经销商与进口商都必须获得MDEL许可证。 加拿大器械许可证(MDL)
我公司办理:ISO13485认证,CE评估报告编写 等产品出口的相关认证
http://sungofda.cn.b2b168.com










