周期4周
品牌SUNGO
公司SUNGO
流程SUNGO
国家欧洲
非级医用防护服、隔离衣、手术衣在MDR中属于普通I类产品,不需要公告机构介入,企业只需通过自我符合性声明的途径进行产品注册,但注册前要完成以下工作
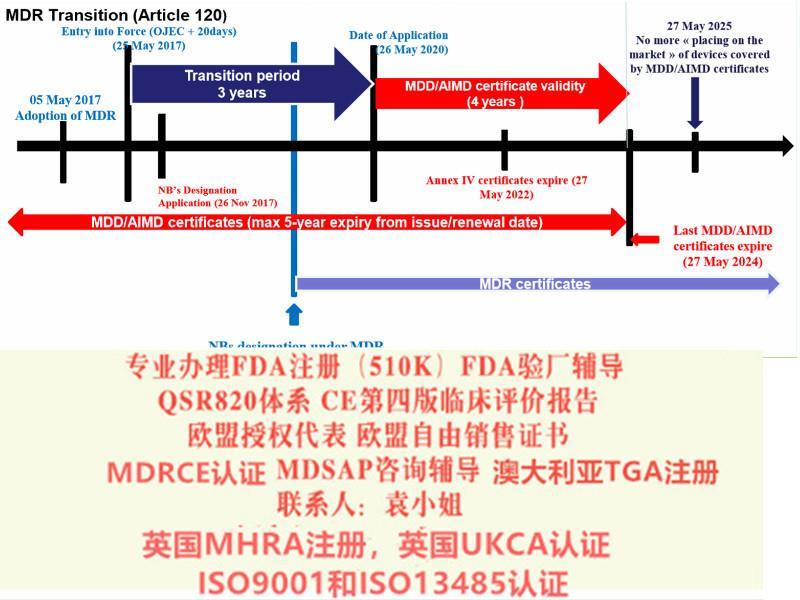
十二、完善评价相关要求 新法规提出: 要求根据Article61和附录XIV partA执行、评估、报告和更新评价资料; 提出对特定III类和IIb类器械,CER中要考虑咨询小组的意见; 对植入和III类器械,提出考虑研究; 要求CER按照PMCF取得数据进行更新; 针对III类和可植入器械,提出了CER更新的频率; 明确实质等同性需考虑的特点; 要求其与风险管理的相互作用 十三、Eudamed数据库 新法规提出: 明确欧洲器械数据库(Eudamed)建立目的和包含的信息(Article 33); 信息的公开性: 要求III类器械和植入式器械,和性能信息通过Eudamed向公众开放。 十四、提出器械的可追溯性(UDI) 除定制和研究器械外,其他器械均需建立UDI系统; UDI信息体现在标签或包装上(不包含集装箱); UDI-DI信息需要载明于符合性声明中(见Article27); Annex VI Part B提出UDI-DI包含的信息; 可植入、重复使用、软件、可配置器械的UDI有要求(见Annex VI Part C) 包装或标签上UDI实施的时间见Article123 (f)。 UDI 发行实体由欧盟会。 过渡性:Article 120指出“在会根据第27(2)条发行实体前,GS1、HIBCC和ICCBBA应被视为的发行实体”。

公司提供的方案包括使用清洁剂清洗,超声波清洗工艺,湿热工艺的验证。 需要办理以下认证可以随时找我 : 1:出口欧盟:MDR CE认证/IVDRCE认证,欧盟授权代表,欧盟注册,欧盟自由销售证书 2:出口英国:英国代表,英国MHRA注册,UKCA认证,英国自由销售证书 3:出口美国:美国FDA注册,FDA510K,QSR820体系 4:中国:国内的器械注册证和生产许可证 5:出口加拿大:加拿大的MDEL注册 6:质量管理体系认证:ISO13485咨询和认证

我公司办理产品出口欧盟、美国、中东南美等的各种认证:
TUV莱茵,TUV南德,SGS等CE认证,CE技术文件编订, CE第四版评价(MEDDEV 2.7.1 Rev 4)编写,欧盟授权代表,欧盟自由销售证书,ISO13485:二016,器械单一审核程序(MDSAP)审核、美国FDA注册(含FDA510K申请), FDA QSR820验厂及整改,FDA警告信应对&RED LIST REMOVAL/QSR820体系/OTC验厂及整改,英国BRC认证咨询,BSCI验厂;口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试、防护服type5/6 (PPE认证咨询(89/686/EC个人防护指令)。
CE第4版评价怎么做?
欧盟第四版评价,您准备好了吗?
已经拿了CE 证书的企业,看过来!
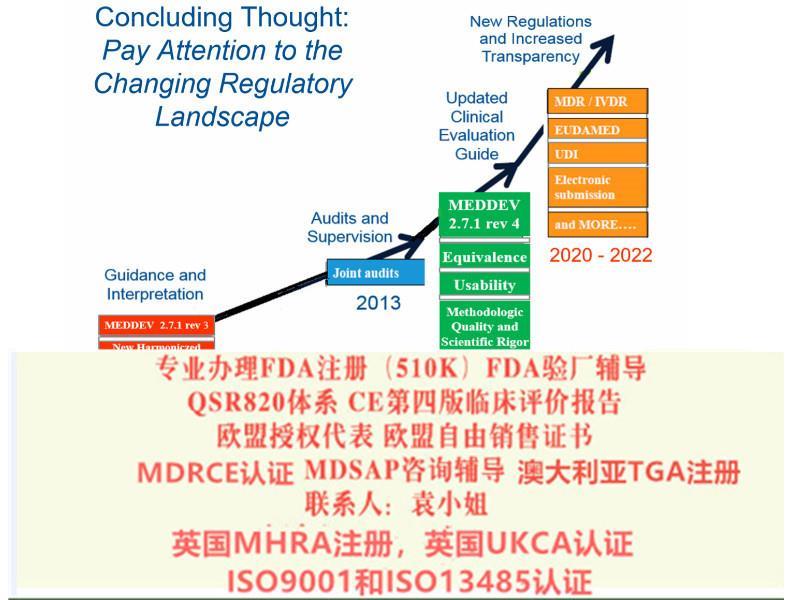
2016年6月,欧盟会发布了有关评价的更新文件MEDDEV 2.7/1 rev. 4.0。与第三版相比,新版增加了对评价的要求。2016年6月发布后即刻生效,没有过渡期。
CE第4版评价怎么做?
公告机构们于今年也在紧锣密鼓地加强审核!已经有不少企业因此被罚红牌。我司应接不暇, 特此布告器械制造商, 必须尽快做出回应,及时更新您们的评价报告(CER) 和质量管理体系(QMS)流程,以符合第四版MEDDEV 2.7/1 rev. 4.0 的要求。
实施计划
CE第4版评价怎么做?
对于第四版的执行时间,各家公告机构做法有所不同, 所持的意见也不尽相同。据我们所了解,公告机构的基本思路可以简单归纳为:
-高风险产品和植入器械(例如Class III 和Class IIb), 公告机构期望器械制造商马上执行。
-低风险器械(如 Class IIa, Is, Im), 公告机构会适当放宽期限,有些机构要求明年内完成更新; 而有些机构则放宽对state of the art 的要求。
应对措施
制造商应该对第四版进行差距分析,从而:
1)对QMS(质量管理体系)的流程进行影响分析;
2)对CER(现有评价报告)进行差距分析;
3)对实际更新准备过渡计划(过渡计划应考虑与产品相关风险及证书到期日)。
如何更新CER
-上市后监督信息(PMS & PMCF)
-当前技术水平 (State of the art)
重要信息
PMCF 是强制的,这也是新欧盟法规MDR **的重要内容之一。对于下列情形,器械生产商需要做好充分准备:
1)之前的CER (上市评价) 走的是等同性路径(特别是高风险产品,如:Class III 和植入器械)
2)产品使用的风险高
3)针对高风险的解剖部位/ 高风险的人群
4)出现了有关性和有效性方面的新的信息
5)创新的器械
6)器械的设计适应症和预期用途发生重大的变化
MEDDEV 2.7.1 Rev4主要变化:
1主要变化之一:报告的更新频率
按照新版报告指南的要求,对于高风险或者新设备,应每年更新;对于低风险的设备,每2-5年更新。对于如何确定更新的频率需要有定义。对于任何风险类别的器械,如果从PMS收集到的信息影响到评价或者结论,CER需要进行更新。
2主要变化之二:报告编写人和评价人的资质
按照新版报告指南的要求,对于报告的编写人提出了资质要求。包括需要有相关的高等教育学位以及至少五年的经历,或者十年的工作经历,如果学位不够的话。如果不能满足要求,需要对其资质进行判定和说明。
3 主要变化之三:评价报告需要有明确的可测量的目标
第四版评估指南对于评估报告的目的有更明确的描述,需要与器械的性、性能以及风险?收益平衡进行更加清晰和详细的描述,在指南的第7部分和附件5中有详细的描述。
4主要变化之四:确定技术发展水平
第四版评估指南对于设备的技术发展水平和处理方式的建立和文件化,提供了更加详细的描述。这包括确定设备的性和性能,以及被宣称的比对器械,行业的基准器械,或者其他的类似器械;需要包括风险和收益的分析。
5 主要变化之五:数据的科学性和有效性
第四版评估指南非常强调数据的科学性和有效性,包括从统计学考虑。这贯穿于整个指南文件规定的阶段,包括影响数据完整性的因素,数据的客观性和权重,文件搜集的方法,数据的评估和权重,数据是否阐述了符合性的分析。
我公司办理:出口美国需要FDA注册,FDA510K,美国代理人(SUNGO可以做510K以及满足FDA要求的510K检测报告,7月份签约价格有优惠)
http://sungofda.cn.b2b168.com