周期4周
品牌SUNGO
公司SUNGO
流程SUNGO
国家欧洲
欧盟授权代表的名称和地址必须器械随附的信息上,例如(包装)标签和使用说明。如果发生事故,欧盟授权代表将协助并协调向主管当局报告事件。
我司可以办理: 1:欧盟MDR CE,欧盟授权代表,欧盟注册 2:美国FDA注册,FDA510K 3:国内的器械注册证和生产许可证 4:加拿大的MDEL注册 5:ISO13485咨询和认证

制造商的义务经销商,制造商,进口商等的义务及其关系现已在该条例中明确规定。*10条制造商应有风险管理制度(*2款)和质量管理制度(*9款);进行评估(*3段);编写技术文件(*4段);,并采用合格评定程序(*6段)。制造商亦须对其产品在市场上销售后负责(*12、13、14段)。它们必须有适当的制度来弥补它们对有缺陷的装置造成的损害所负的财务责任(*16段)。每个制造商应一名负责合规的人员(*15条)。一些可植入设备的制造商必须为患者提供植入卡(*18条)。一旦制造商完成所有这些义务,他们应制定一份符合性声明(*19条),并在其设备上应用CE标记(*20条)。欧盟/欧洲经济区以外的制造商应与欧盟/欧洲经济区内的授权代表签订合同(*11条)。授权代表(*11条)、进口商(*13条)和分销商(*14条)的义务也作了明确说明。公告机构NB根据MDR新规定,必须公告机构。器械将被要求满足更严格的标准,特别是在能力方面。

ce*四版评价报告
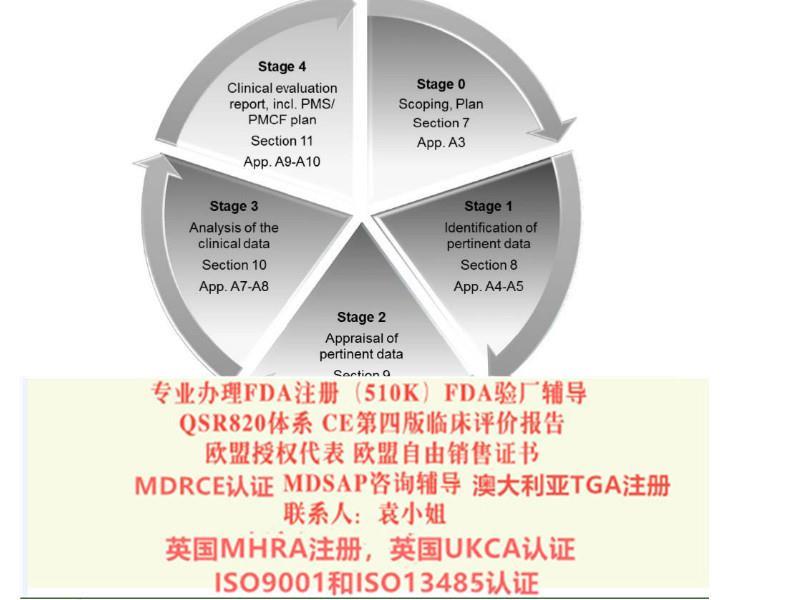
《评价指南MEDDEV 2.7.1 *三版》(以下简称“*三版”)要求制造商记录CER的目的和范围,根据基本要求确定产品的性、性能和风险结点,但其范围和风险结点之间的联系并未在附录F公告机构评价检查表中说明。而*四版更加明确了CER的目的要结合产品性、性能和风险/受益情况,具体在*7节和附录5中有详细的指南。
5.确定被认可的水平(State of Art)
条款8.2提供了更多关于明确和记录被认可(State of Art)和已有方案的详细说明。包括明确产品的性和性能、产品等同性申明、任何基本产品或其他类似产品,以及其他已有方案的风险和受益分析。
6.数据的科学和有效性
*四版更强调数据的有效性,包括数据统计技术的应用。条款9.3.1(“如何评价方法学质量和科学有效性”)强调了影响不同类型数据有效性的因素。此外,贯穿整个器械生命周期的评价指南更为条分缕析:其中罗列了可能影响数据完整性、客观性或加权方法的因素,包括文献检索和检索方法(*8节和附录5),数据评估和加权(*9节和附录6),数据分析和证实符合性(*10节和附录7)。
7.等同性
在*三版中等同性仅仅是附录F中的一个脚注,而在*四版附录1中等同性的要求有了更详细的说明。等同性的标准(、技术、生物学)没有改变,但是如何记录以及影响等同性的因素在*四版中有了更详细的说明。*四版特别要求应详细说明设计差异及其对性和性能的影响,应提供比较图纸和图表,并要求每一个产品的等同性声称必须满足所有三个等价性标准要求。
8.授权查看等同性产品数据
*四版也要求了公告机构需评估制造商等同性产品的数据(附录 A12.2.3);制造商需在合同中写明,允许公告机构评估等同性申明中其竞争对手的同类产品数据,这将是法规的一个转变点。
9.何时需要进行试验
附录2介绍了针对不同风险类型的产品进行试验的关键考量因素,以及制造商应该如何确定他们是否有足够的证据。
10.风险/受益的评价
附录7说明了如何通过数据分析产品的性和性能。附录7.2特别讨论了产品的风险/受益情况,包括风险和受益的评价和量化,以及整体风险/受益情况的评价。*四版中细化了产品上市后的数据价值,以及影响数据评价有效性的因素。
11.上市后监督(PMS)和上市后跟踪(PMCF)
*四版指南文件,强化了评价、PMS和PMCF之间的关系。附录12强调公告机构要确保制造商的PMCF已建立或者恰当的免除,且CER中记录的检索数据和结论是合理的。
器械指南MEDDEV 2.7.1*四版变更内容-评价
一、综述
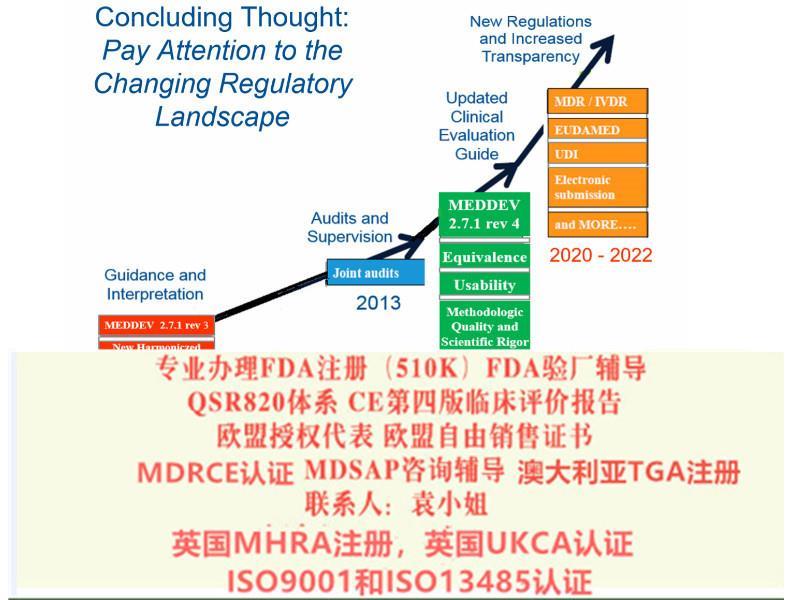
本指南为CE-器械指令应用问题相关的指南中的一部分,是关于评价资料撰写的一些原则,在MEDDEV 2.7.1 *三版的基础上增加了一部分内容,总体变更内容如下:
内容更多、更详细
提供更多有益的和案例
明确了现有要求,而非只是介绍
对于制造商应如何进行一个健全、系统的评价,以及如何数据和结论的科学有效性,有了更明确的
结合了欧盟器械法规(MDR),我们相信这将帮助器械制造商应对从指令到法规的过渡
二、
主要变化内容
1.评价报告(CER)的更新频率
条款6.2.3要求,对于高风险或不成熟的产品,CER应至少每年更新一次;对于低风险且比较成熟的产品,CER应每2至5年更新一次。CER更新的频率也需要有合理说明。对于所有分类下的产品,当产品上市后监督收集的数据会影响评价或结论时,CER必须更新。
2.评价报告撰写者和评价者的资质要求
条款6.4对评价报告撰写者和评价者的背景和经验有明确的要求,包括需要相关的高等*和五年相关经验;若高等*非工作的先决条件,则要求具备10年相关的经验。与相关要求产生的偏离应记录在案,并有正当理由。从现在起,所有评价者必须做出利益关系申明。
3.Abbreviations 缩写词
明确给出了器械中常用名词的英文缩写。
AIMDD: Active implantable medical device directive (Council Directive 90/385/EEC amended by Directive 2007/47/EC) 有缘植入器械
CEAR: Clinical Evaluation Assessment Report 评价评估报告
CER: Clinical Evaluation Report 评价报告
ER: Essential Requirement 基本要求
IFU: Instructions For Use 使用说明书
MDD: Medical Device Directive (Council Directive 93/42/EEC amended by Directive 2007/47/EC) 器械指令
说到这里 企业应该如何面对新法规的升级呢?按照MDR法规要求。关键的内容包括如下几个方面: 企业的质量管理体系 EN ISO13485:2016 ,产品的型式试验 TYPE TESTING , 产品的技术文件 TECHNICAL CONSTRUCTION FILES要满足这些要求,通常需要咨询机构和咨询师的协助。本公司拥有一支经验丰富的咨询师队伍可以为贵司提供这些服务,包括: 协助贵司建立/升级器械质量管理体系,将MDR法规的内容整合进去 协助贵司确定产品的欧盟协调标准,确认检测实验室的,样品准备以及检测不合格整改的研讨 按照MDR要求协助贵司准备技术文件,包括风险分析报告,评估资料,基本要求检查表等 协助贵司按照欧盟的要求修订说明书和标签,使其满足出口的要求。
ISO13485认证,CE评估报告编写,FDAQSR820等产品出口的相关认证
http://sungofda.cn.b2b168.com