参数2
流程1
指令3
费用1
周期4
新法规主要在以下几点上发生了变化:
1.器械的定义;
2.器械的分类;
3.基本*和性能要求;
4.技术文件要求;
5.评价;
6.上市后;
7.Eudamed数据库;
8.对NB公告机构的要求(新法规生效后NB将按照新的资质要求重新进行授权);
9.对高风险器械的新增了要求;
我公司办理产品出口欧盟、美国、中东南美等的各种认证:
TUV莱茵,TUV南德等CE认证(MDD/MDR法规),CE技术文件编订, CE第四版评价(MEDDEV 2.7.1 Rev 4)编写,欧盟授权代表,欧盟自由销售证书,ISO13485:二016,器械单一审核程序(MDSAP)审核、美国FDA注册(含FDA510K申请),FDA美国代理人服务/ FDA QSR820验厂及整改,FDA警告信应对&RED LIST REMOVAL/QSR820体系/OTC验厂及整改,英国BRC认证咨询,BSCI验厂;口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试、防护服type5/6 (PPE认证咨询(89/686/EC个人防护指令)。
Q:MDR何时生效?
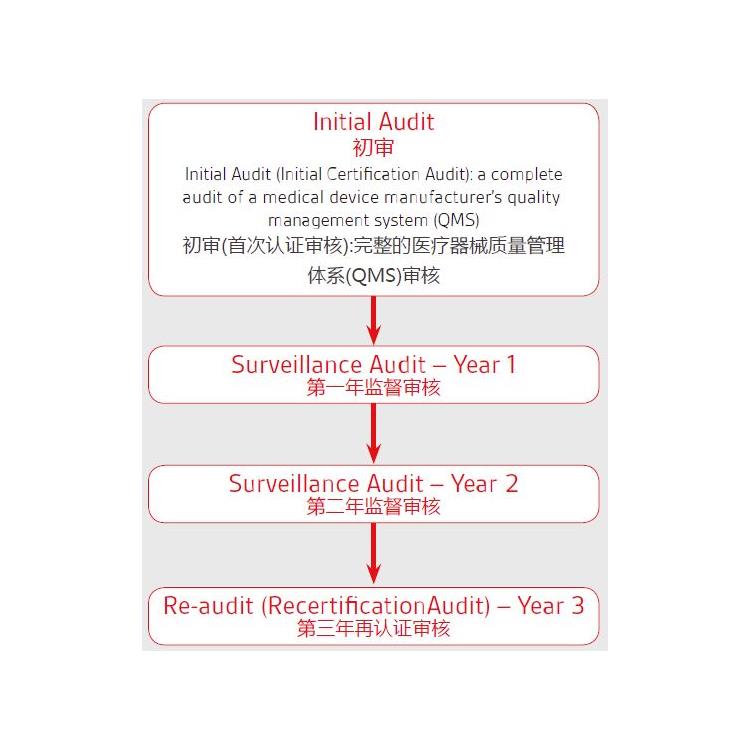
A:2019年5月5日,欧盟正式发布了欧盟器械法规(MDR)。2019年5月25日,MDR正式生效。
器械指令MDD(93/42/EEC)和有源植入类器械指令AIMDD(90/385/EEC)被器械法规MDR(EU 2019/745)取代,法规过渡期为3年。
制造商应在过渡期内更新技术文件和流程以满足法规要求。具体可以参阅法规Article 120中若干过渡条款的要求。
Q:MDR的新要求是否可以延后执行?
A:不可以, MDR覆盖的所有产品都必须满足新法规的要求。过渡期结束后,不符合MDR要求的产品不可在欧盟上市。
Q:证书未在过渡期结束前签发怎么办?
A:对于持有符合MDD或AIMDD的CE证书的制造商,将有3年的过渡期来满足MDR的要求。
在过渡期按照MDD和AIMDD签发的CE证书,在正式生效日期(2020年5月26日)后将继续有效,但有效期多不能**过4年。而在过渡期结束后的证书有效性取决于法规Article 120 clause 3所述规定。
如果您符合MDD的CE证书在过渡期内失效,且又未在过渡期内取得符合MDR的CE证书。那么你的产品必须从欧盟市场退出,直到产品获得符合MDR的CE证书才可重新上市。
Q:公告机构何时开始按照MDR进行符合性评审?
A:所有公告机构需要获得欧盟主管当局的认可后,才能按照MDR进行审核。
Q:.对于QMS是否符合MDR的要求,有具体的截止日期嘛?
A:无论是已满足器械指令(MDD)要求的器械,还是未满足MDD的器械,按照器械法规(MDR)认证都需要符合MDR的要求。
MDR要求制造商能够展现出有效的质量管理体系。因此,要满足MDR的认证要求,您必须按照法规Article 120的要求,在过渡期内建立合格的质量管理体系。
Q:新MDR对分包方(contract manufacturers)有什么影响?
A:如果分包方要为他们所服务的制造商承担责任,那么他们必须遵守法规要求。
如果分包方不需要为他们所服务的制造商承担责任,那么他们仅需代表制造商接受审核,包括飞行检查。
我公司办理产品出口欧盟、美国、中东南美等的各种认证:
TUV莱茵,TUV南德等CE认证(MDD/MDR法规),CE技术文件编订, CE第四版评价(MEDDEV 2.7.1 Rev 4)编写,欧盟授权代表,欧盟自由销售证书,ISO13485:二016,器械单一审核程序(MDSAP)审核、美国FDA注册(含FDA510K申请),FDA美国代理人服务/ FDA QSR820验厂及整改,FDA警告信应对&RED LIST REMOVAL/QSR820体系/OTC验厂及整改,英国BRC认证咨询,BSCI验厂;口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试、防护服type5/6 (PPE认证咨询(89/686/EC个人防护指令)。
英国脱欧后,英国局签发的自由销售证书还有用吗?
要回答这个问题,我们需要弄明白这个自由销售证书的用途。自由销售证书是为了向第三方其打算采购的产品已经获得了在证书出具国的销售,满足了其所有的法规要求。从这个意义上讲,出具国的主管机构在国际上越,声望越高,其证书的效力也越高。英国局目前在国际上是与美国FDA影响力旗鼓相当的主管机构,特别是借助其语言优势和法规完善性,英国局的证书效力和影响力不会因为脱欧有任何影响。
证书为什么需要做海牙认证或者认证?
部分要求证书进行海牙认证或者认证之后才能够在当地使用,这是为了确保文件的真实性而进行的动作。海牙认证的前提是需要目的国和签发国都是海牙公约国的成员才可以实施,必须由英国相关部门完成。例如英国签发的证书,英国是海牙公约国成员,因此只要目的国是海牙成员,例如阿根廷,即可进行海牙认证。如果目的国不是海牙公约国,例如沙特或者埃及,则需要进行认证。认证由目的国驻签发国的大完成。

自由销售证书(出口销售书)用途
一、在收货方海关清关中使用:执行贸易保护的海关要求必须出具出口销售证书、自由销售证书才能清关提货。
二、在进口国注册登记使用:进口方在本国分销销售货物产品时,出于对产品本身的、质量等考虑,要求出具该产品的自由销售证书并在当地质量、门注册登记后才可以在进口国自由销售该批货物。
三、对产品质量是否合格、产品是否合法生产销售的:比如向贸易方以及贸易国:该产品为质量、产品达到相关标准执行什么指令、产品为合法生产销售等其它
四、其它用途,如顾客提出或进口商提出。
自由销售证书
自由销售证书也叫出口销售书 英文名称为:Free Sales Certificate、Certificate of Free Sale或者Certificate For Exportation of Medical Products;简称:FSC 或 CFS。
自由销售证书源于欧洲, 起初,欧洲经济区协定(EEA)个别成员国以及成员国境内的民间协会机构为了促销本国的产品到EEA的第三国,为当地制造商出具自由销售证书,其内容是是产品满足相关标准,满足相关的指令要求,产品可靠,质量达到相关要求,可以在本国本地范围内自由销售,并允许出口之类的。初的自由销售证书由欧洲民间协会、商会等机构出具,后经欧洲一些完善形成一套适合自己的体系。源于欧洲的这种自由销售贸易壁垒,逐渐被世界部分,他们在进口产品时会要求货物发货方提供相应的自由销售证书。
需要出具欧盟自由销售证书,被欧盟指令授权并有的机构应该为EEA成员国的对器械的主管机关Competent Authorities (CAs),多为机构 (比向阿根廷出口的自由销售),而不应该由民间的制造商协会出具。
有了CE标志并进行了相关指令中要求的欧盟注册后,中国的制造商出口欧盟不需要自由销售证书。
产品在欧盟卖,只要有CE证书即可。但您持CE证书去很多非欧盟注册,很多会要求您提供欧盟签发的自由销售。

我公司办理产品出口欧盟、美国、中东南美等的各种认证:
TUV莱茵,TUV南德等CE认证(MDD/MDR法规),CE技术文件编订, CE第四版评价(MEDDEV 2.7.1 Rev 4)编写,欧盟授权代表,欧盟自由销售证书,ISO13485:二016,器械单一审核程序(MDSAP)审核、美国FDA注册(含FDA510K申请),FDA美国代理人服务/ FDA QSR820验厂及整改,FDA警告信应对&RED LIST REMOVAL/QSR820体系/OTC验厂及整改,英国BRC认证咨询,BSCI验厂;口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试、防护服type5/6 (PPE认证咨询(89/686/EC个人防护指令)。
欧盟授权代表在欧盟的产品指令(Directive)英文版里使用的标准术语为(英国式英语) European Authorised Representative, 因为需要欧盟授权代表的多数为位于欧盟的的制造商,尤其以美国的制造商为多, 美国的制造商更喜欢将欧盟授权代表以美式英语书写为: European Authorized Representative。
鉴于中文并非欧盟的语言,因此European Authorised Representative 并没有对应的中文的术语。在中文的翻译里,通常将European Authorised Representative 或European Authorized Representative译为: 欧盟授权代表。 也有翻译为: 欧盟授权代理、欧洲授权代表、欧洲授权代理等。通常简称为:欧盟代表或欧代。也有使用:欧盟代理、欧洲代表、欧洲代理等。在中国台湾,还有使用欧体授权代表或欧体代表。
c) 欧盟制造商的产品在欧盟出现任何故障/事故/召回等问题,应由欧盟授权代表进行联络,通报,并与主管机关进行沟通联系。
如何选择一个的欧盟授权代表
a) 选择有资质,有能力的第三方欧盟授权代表公司。
欧盟境内注册的合法公司/拥有的技术人员,熟悉欧盟相关法规,能帮助制造方解决争端/避免空壳公司,代理商以及展会服务商。
------以上几点可通过查询其欧盟注册证书,拨打其欧盟境内的电话看是否为电话录音等途径进行确认。
b) 签订有效的欧盟授权代表协议或合同。
合同/协议的甲乙双方的名称和地址必须和将来加贴CE标志的产品的包装/标签上的制造商和欧盟授权代表的名称和地址完全一致。
c) 应注意以下事项:
欧盟授权代表合同条款应是欧盟的主要语言版本,一般为欧代所在的语言版本。
避免在欧盟境内无固定办公地点和固定联络电话的欧盟授权代表。
避免经销商兼任欧盟授权代表。
避免在欧洲留学的亲戚朋友兼任欧盟授权代表。
针对制造商的MDR合规性要求
III级高风险有源植入式CMD也需要获得公告机构的质量管理体系(QMS)认证合格评定程序。这些设备的QMS证书也必须输入到Eudamed设备数据库中。
http://sungofda.cn.b2b168.com