我公司专业办理医疗产品出口欧盟、美国、中东南美等国家的各种认证:
TUV莱茵,TUV南德,***等CE认证,全套CE技术文件编订, CE第四版临床评价(MEDDEV 2.7.1 Rev 4)编写,欧盟授权代表,欧盟自由销售证书,ISO13485:二016,医疗器械单一审核程序(MDSAP)审核、美国FDA注册(含FDA510K申请), FDA QSR820验厂辅导及整改,FDA警告信应对&RED LIST REMOVAL/QSR820体系辅导/OTC验厂辅导及整改,英国BRC认证咨询,BSCI验厂辅导;口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试、防护服type5/6 (PPE认证咨询(89/686/EC个人防护指令辅导)。
我公司临床报告业务优势
按照第四版临床报告指南的要求,对于临床评估报告的撰写人资格有相应的要求。SUNGO组建了临床评估业务*技术小组,包括医学博士,国际**认证机构评审人员,世界**医疗器械企业质量经理等相关专业人员。目前SUNGO已经交付了近百种产品的临床评估报告,其中包括外科手术导航系统,骨科植入产品等较高风险和复杂程度的产品。
我司将基于目前申请企业的现状,依据MEDDEV 2.7.1 Rev 4的相关要求,编制能够符合发证机构发证公司的全英文临床评估报告。
针对MEDDEV2.7.1 Rev 4,我司可以协助您:
1、协助建立临床评价程序;
2、建立临床评价方案;
3、寻找等同产品,进行等同分析;
4、搜索文献及其他临床数据;
5、临床数据分析;
6、完成临床评价报告;
7、全英文临床评估报告;
8、认证机构审核通过。
背景
MED 2_7_1 Rev 4的发布对临床评估报告提出了新的要求,所有打算申请CE认证或者已经取得CE认证需要继续维持证书有效的所有企业,都需要按照新版指南更新临床评估报告。
新版指南的变化
相对于之前的临床报告,第四版的临床报告主要变化体系在:
1.临床报告更新的频率
2.报告编写人和评价人的资质
3.评估报告需要有明确的可测量目标
4.确定技术发展水平
5.数据的科学性和有效性
6.比对器械
7.比对器械的数据获得
8.什么时候需要临床试验
9.售后监督和售后临床跟踪
10.风险?收益。
一次性无菌注射器带针、一次性无菌输液器带针、电子体温计、医用润滑剂、活组织检查针、输液泵、雾化器、一次性麻醉穿刺针等产品,TUV南德/TUV莱茵/***/BSI/KIWA等公告机构要求的欧盟第四版医疗器械临床评价/评估报告,提供编写或更新。
此外,MEDDEV 2.7.1第四版还规定了公告机构的角色和职责。一些主要变化是,公告机构必须为临床评价报告的评估建立所要求的QMS程序,且必须拥有评估临床评价报告所需的专业知识。除此之外,公告机构还需要对其评估的所有临床评价案例出具临床评价评估报告(CEAR)。临床评价评估报告(CEAR)可作为设计卷宗或技术文档报告(如有)的一部分。
总之,MEDDEV 2.7.1 第四版将导致更多的临床试验以及可能更大的样本量,相应地,公告机构则需更加严格的审查所有适用的基本要求 (包括那些与可用性相关的基本要求) 是否已满足。临床评价报告本身也需要更频繁的更新、由更*的评价者来编写和审核、且与器械生命周期的各个阶段更紧密的结合。随着MEDDEV 2.7.1第四版的出台且没有新旧版本的过渡期,制造商利益的做法便是从现在开始与公告机构讨论如何开始实施这些新要求并从现在开始执行差距评估和资源需求评估。差距评估已迫在眉睫,因为制造商需要尽快为额外的临床数据进行预算。
欧盟医疗器械临床评价指南于进行了第四次修订 MEDDEV 2.7.1第四版的变化之一,就是证明“实质等同”的难度要比之前大很多。关键的要求就是一个器械必须满足所有三个一般标准(临床、技术和生物)才能证明“实质等同”。另外,MEDDEV 2.7.1第四版还要求制造商在器械的技术文档中包含对比器械的支持性非临床信息(如临床前报告),也就是说,制造商必须能够完全获取(也就是“拥有”)对比器械的技术文档/设计卷宗。在临床评价过程中,采用的相关临床数据必须来源于符合医疗器械指令MDD/有源植入性医疗器械指令AIMDD要求的医疗器械,认识到这一点很重要。因此,如果选择非CE认证器械(如:获得美国510K或PMA的器械)作为实质等同器械(对比器械),那么制造商必须就器械批准上市的国家/地区和欧盟之间在患者人群或临床实践上的任何差异作出合理解释。
MEDDEV 2.7.1第四版的附录2就何时需要进行临床试验提供了指南。当然,根据医疗器械指令93/42/EEC附录X的1.1a章节,可植入器械和III类器械必须进行临床试验(除非有其它正当理由)。除此之外,MEDDEV 2.7.1第四版还规定了以下情形也需要进行临床试验:器械运用了新技术或现有技术的临床新用途;现有的临床数据存在任何差距不足以证明器械(包括I类、IIa类和IIb类器械)使用的受益、风险、要求或*满足适用的基本要求。很明显,以上规定使得很少有制造商能够仅仅通过临床文献和临床经验数据来证明器械满足适用的基本要求。
关于MEDDEV2.7.1 Rev 4,SUNGO可以协助您:
1、协助建立临床评价程序;
2、建立临床评价方案
3、寻找等同产品,进行等同分析;
4、搜索文献及其他临床数据;
5、临床数据分析;
6、完成临床评价报告。
CE临床评价
我公司临床报告业务优势
按照第四版临床报告指南的要求,对于临床评估报告的撰写人资格有相应的要求。SUNGO组建了临床评估业务*技术小组,包括医学博士,国际**认证机构评审人员,世界**医疗器械企业质量经理等相关专业人员。目前SUNGO已经交付了近百种产品的临床评估报告,其中包括外科手术导航系统,骨科植入产品等较高风险和复杂程度的产品。
我司将基于目前申请企业的现状,依据MEDDEV 2.7.1 Rev 4的相关要求,编制能够符合发证机构发证公司的全英文临床评估报告。

器械临床评价指南MEDDEV 2.7.1第四版已于2016年6月出炉。
在各主管当局对公告机构的联合审查愈发严格的大背景下,此次MEDDEV 2.7.1的修订应运而生,使得公告机构对临床评价审核的严格程度上了一个新的台阶。正因为这些要求之前没有写进法规,公告机构便忽略了对这些隐形要求的评审/审查,这也推动了这次MEDDEV 2.7.1的修订。
实际上,这次MEDDEV 2.7.1的修订应该说是一次全新的改写,包含了许多新的附录和指南。新版的MEDDEV 2.7.1更具指导意义,在对比器械的证据使用方面,新版的MEDDEV 2.7.1也更加规范。在5月底的新器械法规(MDR)的讨论会上所达成的共识方面,新版的MEDDEV 2.7.1并没有体现即将出现在新器械法规中的变化的要求。一旦新器械法规终发布,器械*组(MDEG)的临床工作组便会考虑进一步修订MEDDEV 2.7.1来满足新的法规要求。
此次的全新改写带来了诸多变化,其中的一些变化对制造商具有重大影响。
CE新版临床评价报告
为加强对临床评价的评审,“目标用户的可用性”现已明确列入MEDDEV 2.7.1第四版,成为评价者需要考虑的因素。该变化可能导致的结果就是需要有更多有关性能测试和数据的评判性评价来支持可用性。
另外,“贯穿整个器械生命周期的临床评价”成为了MEDDEV 2.7.1第四版的一个关键主题。在开发阶段,器械的研发由临床评价和风险管理指导。临床评价可能会用来定义有关器械临床*性和性能的需求,以及评估存在的数据和数据中的任何差异(这些数据和差异可能需要通过临床研究获得)。对于CE认证申请,需求递交临床评价报告(CER)来证明有“充分的临床证据”(MEDDEV 2.7.1第四版中的新定义)证明器械符合涵盖临床性能和临床*性的基本要求,以及识别需要通过上市后监督(PMS)来解决的任何问题。在上市后阶段,需要对上市后监督数据持续评审来不断确认器械的风险/受益情况、临床*性和性能。这些数据需要及时输入临床评价过程。
新版 MEDDEV 2.7.1 还对临床评价报告的更新频率进行了规定。制造商必须基于器械的 “重大风险” 以及 “成熟程度” (这两个新概念在新版 MEDDEV 2.7.1 中有描述) 明确临床评价报告的更新频率并说明理由。MEDDEV 2.7.1第四版规定, 若器械存在重大风险或不太成熟,其临床评价报告必须至少每年更新一次;若器械不存在重大风险且比较成熟,必须每2到5年更新一次。制造商必须为其规定的更新频率说明理由并在证书到期换证时配合公告机构进行更新。
对于大多数企业来说,这是一个重大的变化,直接导致企业改变之前的方法来对上市后监督和临床数据进行持续不断的评估。与此同时,随着持续改进的元素纳入到法规科学之中,预计需要大量增加资源。
在MEDDEV 2.7.1第四版中,对临床数据评价者的资质有了更严格的要求。除了第三版中的相关规定外,第四版还规定评价者应具备临床研究设计和生物统计的相关知识、熟知相关法规要求且有医学写作的相关经验。此外,评价者还必须拥有高等学位以及5年有案可查的专业经验,或者,若给定的任务不需要高等学位,则要求评价者拥有10年有案可查的专业经验。如果制造商能够用文件证明与上述规定的偏离是合理的,MEDDEV 2.7.1第四版也有相关的例外条款。
CE第4版临床评价报告背景
MED 2_7_1 Rev 4的发布对临床评估报告提出了新的要求,所有打算申请CE认证或者已经取得CE认证需要继续维持证书有效的所有企业,都需要按照新版指南更新临床评估报告。
新版指南的变化
相对于之前的临床报告,第四版的临床报告主要变化体系在:
1.临床报告更新的频率
2.报告编写人和评价人的资质
3.评估报告需要有明确的可测量目标
4.确定技术发展水平
5.数据的科学性和有效性
6.比对器械
7.比对器械的数据获得
8.什么时候需要临床试验
9.售后监督和售后临床跟踪
10.风险/收益。
我公司临床报告业务优势
按照第四版临床报告指南的要求,对于临床评估报告的撰写人资格有相应的要求。SUNGO组建了临床评估业务*技术小组,包括医学博士,国际**认证机构评审人员,世界**器械企业质量经理等相关专业人员。目前SUNGO已经交付了近百种产品的临床评估报告,其中包括手术导航系统,骨科植入产品等较高风险和复杂程度的产品。
我司将基于目前申请企业的现状,依据MEDDEV 2.7.1 Rev 4的相关要求,编制能够符合发证机构发证公司的全英文临床评估报告。
针对MEDDEV2.7.1 Rev 4,我司可以协助您:
1、协助建立临床评价程序;
2、建立临床评价方案;
3、寻找等同产品,进行等同分析;
4、搜索文献及其他临床数据;
5、临床数据分析;
6、完成临床评价报告;
7、全英文临床评估报告;
8、认证机构审核通过。
我司将基于目前申请企业的现状,依据MEDDEV 2.7.1 Rev 4的相关要求,编制能够符合发证机构发证公司的全英文临床评估报告。

出口:美国FDA注册(含FDA510K申请)、 FDA QSR820验厂及整改、FDA警告信应对&RED LIST REMOVAL/QSR820体系/OTC验厂及整改、CE认证(CE整套技术文件编订、 CE第四版临床评价(MEDDEV 2.7.1 Rev 4)编写)、ISO13485:2016、欧盟授权代表、欧盟销售证书、英国BRC认证,BSCI验厂;口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试、防护服type5/6 (PPE认证(89/686/EC个人防护指令)。
全套CE技术文件编订,欧盟器械临床评价指南于进行了第四次修订MEDDEV 2.7.1第四版的大变化之一,就是证明“实质等同”的难度要比之前大很多。全套CE技术文件编订,关键的要求就是一个器械必须满足所有三个一般标准(临床、技术和生物)才能证明“实质等同”。另外,MEDDEV 2.7.1第四版还要求制造商在器械的技术文档中包含对比器械的支持性非临床信息(如临床前报告),也就是说,制造商必须能够完全获取(也就是“拥有”)对比器械的技术文档/设计卷宗。
CE第四版临床评价编写,在临床评价过程中,采用的相关临床数据必须来源于符合器械指令MDD/有源植入性器械指令AIMDD要求的器械,认识到这一点很重要。CE第四版临床评价编写,如果选择非CE认证器械(如:获得美国510K或P的器械)作为实质等同器械(对比器械),CE第四版临床评价编写,那么制造商必须就器械批准上市的/地区和欧盟之间在患者人群或临床实践上的任何差异作出合理解释。
2.7.1 Rev 4临床评价报告
MEDDEV 2.7.1第四版的附录2就何时需要进行临床试验提供了指南。CE第四版临床评价编写,当然,根据器械指令93/42/EEC附录X的1.1a章节,可植入器械和III类器械必须进行临床试验(除非有其它正当理由)。CE第四版临床评价编写,MEDDEV 2.7.1第四版还规定了以下情形也需要进行临床试验:器械运用了新技术或现有技术的临床新用途;现有的临床数据存在任何差距不足以证明器械(包括I类、IIa类和IIb类器械)使用的受益、风险、要求或满足适用的基本要求。CE第四版临床评价编写,很明显,以上规定使得很少有制造商能够仅仅通过临床文献和临床经验数据来证明器械满足适用的基本要求。
此外,MEDDEV 2.7.1第四版还规定了公告机构的角色和职责。CE第四版临床评价编写,一些主要变化是,公告机构必须为临床评价报告的评估建立所要求的QMS程序,且必须拥有评估临床评价报告所需的专业知识。CE第四版临床评价编写,公告机构还需要对其评估的所有临床评价案例出具临床评价评估报告(CEAR)。临床评价评估报告(CEAR)可作为设计卷宗或技术文档报告(如有)的一部分。
总之,MEDDEV 2.7.1 第四版将导致更多的临床试验以及可能更大的样本量,相应地,公告机构则需更加严格的审查所有适用的基本要求 (包括那些与可用性相关的基本要求) 是否已满足。CE第四版临床评价编写,临床评价报告本身也需要更频繁的更新、由更的评价者来编写和审核、且与器械生命周期的各个阶段更紧密的结合
CE第四版临床评价,随着MEDDEV 2.7.1第四版的且没有新旧版本的过渡期,符合制造商大利益的做法便是从现在开始与公告机构讨论如何开始实施这些新要求并从现在开始执行差距评估和资源需求评估。CE第四版临床评价,差距评估已迫在眉睫,因为制造商需要尽快为额外的临床数据进行预算。因此,如果临床试验需要在2019年启动的话,则需要将评估成本记入明年的预算计划内。
CE第四版临床评价,关于MEDDEV2.7.1 Rev 4,我司可以协助您:
1、协助建立临床评价程序;
2、建立临床评价方案
3、寻找等同产品,进行等同分析;
4、搜索文献及其他临床数据;
5、临床数据分析;
6、完成临床评价报告。
CE第四版临床评价,我司可办CE技术文件的整改,CE技术文件的编订,风险管理报告的编写,依据MEDDEV2.7.1Rev4编写的临床评估报告,欧盟代表服务,销售certificateCFS,美国FDA注册,FDA验厂等。CE第四版临床评价,报告编写人和评价人的资质 按照新版临床报告指南的要求,对于临床报告的编写人提出了资质要求。包括需要有相关专业的高等教育学位以及至少五年的专业经历,或者十年的专业工作经历,如果学位不够的话。如果不能满足要求,需要对其资质进行判定和说明。
CE第四版临床评价,临床报告更新的频率 按照新版临床报告指南的要求,对于高风险或者新设备,应每年更新;对于低风险的设备,每2-5年更新。CE第四版临床评价,对于如何确定更新的额频率需要有定义。对于任何风险类别的器械,如果从PMS收集到的信息影响到评价或者结论,CER需要进行更新。

我公司专业办理医疗产品出口欧盟、美国、中东南美等国家的各种认证:
TUV莱茵,TUV南德,***等CE认证,全套CE技术文件编订, CE第四版临床评价(MEDDEV 2.7.1 Rev 4)编写,欧盟授权代表,欧盟自由销售证书,ISO13485:二016,美国FDA注册(含FDA510K申请), FDA QSR820验厂辅导及整改,FDA警告信应对&RED LIST REMOVAL/QSR820体系辅导/OTC验厂辅导及整改,英国BRC认证咨询,BSCI验厂辅导;口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试、防护服type5/6 (PPE认证咨询(89/686/EC个人防护指令辅导)。
CE第四版临床评价
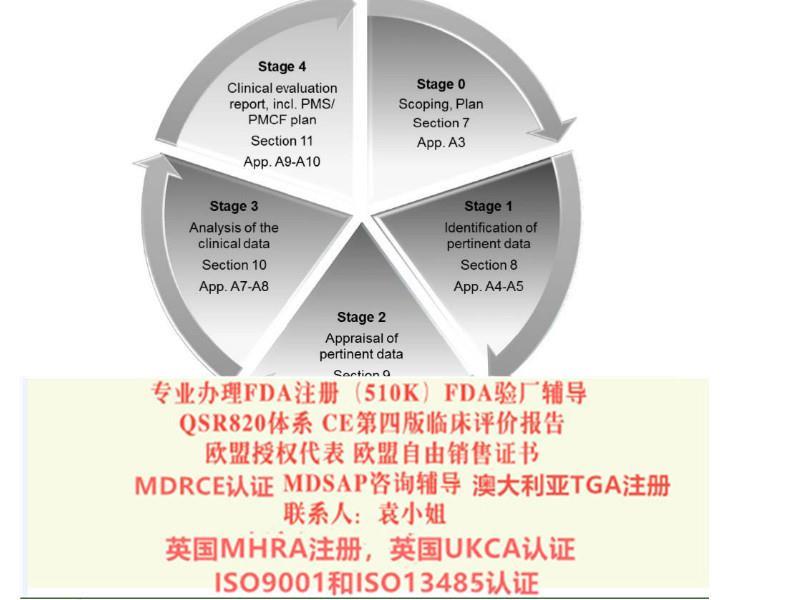
1: 新版指南的变化: 相对于之前的临床报告,第四版的临床报告主要变化体系在:1.临床报告更新的频率,2.报告编写人和评价人的资质,3.评估报告需要有明确的可测量目标,4.确定技术发展水平,5.数据的科学性和有效性,6.比对器械,7.比对器械的数据获得,8.什么时候需要临床试验,9.售后监督和售后临床跟踪,10.风险—收益。
2:关于CE认证的技术文件里第4版临床评价(MEDDEV 2.7.1 Rev 4)的背景:MED 2_7_1 Rev 4的发布对临床评估报告提出了新的要求,所有打算申请CE认证或者已经取得CE认证需要继续维持证书有效的所有企业,都需要按照新版指南更新临床评估报告。
3: 针对MEDDEV2.7.1 Rev 4,我司可以协助您:1、协助建立临床评价程序;2、建立临床评价方案;3、寻找等同产品,进行等同分析;4、搜索文献及其他临床数据;5、临床数据分析;6、完成临床评价报告; 7、全英文临床评估报告;8、认证机构审核通过。
4:我公司临床报告业务优势:按照第四版临床报告指南的要求,对于临床评估报告的撰写人资格有相应的要求。SUNGO组建了临床评估业务*技术小组,包括医学博士,国际**认证机构评审人员,世界**医疗器械企业质量经理等相关专业人员。目前SUNGO已经交付了近百种产品的临床评估报告,其中包括外科手术导航系统,骨科植入产品等较高风险和复杂程度的产品。
我司将基于目前申请企业的现状,依据MEDDEV 2.7.1 Rev 4的相关要求,编制能够符合发证机构发证公司的全英文临床评估报告。
我公司专业办理医疗产品出口欧盟、美国、中东南美等国家的各种认证:
TUV莱茵,TUV南德,***等CE认证,全套CE技术文件编订, CE第四版临床评价(MEDDEV 2.7.1 Rev 4)编写,欧盟授权代表,欧盟自由销售证书,ISO13485:二016,美国FDA注册(含FDA510K申请), FDA QSR820验厂辅导及整改,FDA警告信应对&RED LIST REMOVAL/QSR820体系辅导/OTC验厂辅导及整改,英国BRC认证咨询,BSCI验厂辅导;口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试、防护服type5/6 (PPE认证咨询(89/686/EC个人防护指令辅导)。
1: 新版指南的变化: 相对于之前的临床报告,第四版的临床报告主要变化体系在:1.临床报告更新的频率,2.报告编写人和评价人的资质,3.评估报告需要有明确的可测量目标,4.确定技术发展水平,5.数据的科学性和有效性,6.比对器械,7.比对器械的数据获得,8.什么时候需要临床试验,9.售后监督和售后临床跟踪,10.风险—收益。
2:关于CE认证的技术文件里第4版临床评价(MEDDEV 2.7.1 Rev 4)的背景:MED 2_7_1 Rev 4的发布对临床评估报告提出了新的要求,所有打算申请CE认证或者已经取得CE认证需要继续维持证书有效的所有企业,都需要按照新版指南更新临床评估报告。
3: 针对MEDDEV2.7.1 Rev 4,我司可以协助您:1、协助建立临床评价程序;2、建立临床评价方案;3、寻找等同产品,进行等同分析;4、搜索文献及其他临床数据;5、临床数据分析;6、完成临床评价报告; 7、全英文临床评估报告;8、认证机构审核通过。
4:我公司临床报告业务优势:按照第四版临床报告指南的要求,对于临床评估报告的撰写人资格有相应的要求。SUNGO组建了临床评估业务*技术小组,包括医学博士,国际**认证机构评审人员,世界**医疗器械企业质量经理等相关专业人员。目前SUNGO已经交付了近百种产品的临床评估报告,其中包括外科手术导航系统,骨科植入产品等较高风险和复杂程度的产品。
我司将基于目前申请企业的现状,依据MEDDEV 2.7.1 Rev 4的相关要求,编制能够符合发证机构发证公司的全英文临床评估报告。
-/gjhdff/-
http://sungofda.cn.b2b168.com