我公司专业办理医疗产品出口欧盟、美国、中东南美等国家的各种认证:
TUV莱茵,TUV南德,***等CE认证(MDD/MDR法规),全套CE技术文件编订, CE第四版临床评价(MEDDEV 2.7.1 Rev 4)编写,欧盟授权代表,欧盟自由销售证书,ISO13485:二016,医疗器械单一审核程序(MDSAP)审核、美国FDA注册(含FDA510K申请),FDA美国代理人服务/ FDA QSR820验厂辅导及整改,FDA警告信应对&RED LIST REMOVAL/QSR820体系辅导/OTC验厂辅导及整改,英国BRC认证咨询,BSCI验厂辅导;口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试、防护服type5/6 (PPE认证咨询(89/686/EC个人防护指令辅导)。
1. 什么是自由销售证书?
自由销售证书Free Sales Certificates,简称FSC,自由销售证书,称自由销售证明,英文名称为:Free Sale Certificate ,简称FSC,还可以称出口销售证明。通常用于医疗产品的出口销售,证明该医疗产品在证书出具国可以自由销售。
EEA成员国及其他个别国家境内的机构(有的甚至是当地民间的制造商协会)为了促销本国(本地)制造的产品出口到EEA境外的其他第三国,为当地的制造商出具自由销售证书,其内容是证明其产品满足了相关EC指令的要求,可以在本国(本地)范围内自由销售。需要出具欧盟自由销售证书,一被欧盟指令授权并有资格的机构应该为EEA成员国的医疗器械主管机关Competent Authorities (Cas)。有了CE标志并进行了相关指令中要求的欧盟注册后,中国的制造商出口欧盟不需要自由销售证书,当您持有的CE证书去其他非欧盟国家注册,有些国家**又是会要求您提供欧盟**签发的自由销售证书。
办理自由销售证明的流程:
1. 准备文件(包括CE证书,文件,企业信息等)
2. 由欧代将资料提交到EEA各成员国主管机关(英国为DH)
3. 签发证书
4. 进行**公证
海牙成员国
尔巴尼亚、安道尔、安提瓜和巴布达、阿根廷、亚美尼亚、阿塞拜疆 澳大利亚、奥地利、巴哈马、巴巴多斯、白俄罗斯、比利时 伯利兹、波斯尼亚和黑塞哥维那、博茨瓦纳 文莱、保加利亚 中国香港和中国澳门 哥伦比亚、库克群岛、克罗地亚、塞浦路斯 捷克共和国、丹麦、多米尼克,英联邦的、厄瓜多尔、萨尔瓦多、爱沙尼亚 斐济、芬兰、法国、格鲁吉亚(从2007年5月14日)德国 希腊 格林纳达 洪都拉斯、匈牙利、印度 冰岛、爱尔兰、以色列、意大利、日本 哈萨克斯坦 韩国、拉脱维亚、莱索托 利比里亚 列支敦士登、立陶宛、卢森堡、马其顿、马拉维、马耳他 马绍尔群岛、毛里求斯、墨西哥、摩尔多瓦(从2007年3月16日起) 摩纳哥、黑山、纳米比亚、荷兰、新西兰、纽埃 挪威、巴拿马、波兰、葡萄牙、罗马尼亚、俄罗斯联邦 圣基茨和尼维斯、圣卢西亚、圣文森特和格林纳丁斯、萨摩亚、圣马力诺 塞尔维亚、塞舌尔、斯洛伐克、斯洛文尼亚、南非、西班牙 苏里南、史瓦济兰、瑞典、瑞士、汤加、特里尼达和多巴哥 土耳其、乌克兰、英国、美国、委内瑞拉.

背景
MED 2_7_1 Rev 4的发布对临床评估报告提出了新的要求,所有打算申请CE认证或者已经取得CE认证需要继续维持证书有效的所有企业,都需要按照新版指南更新临床评估报告。
新版指南的变化
新版指南对于临床报告的结构要求
SUNGO 临床报告业务优势
按照第四版临床报告指南的要求,对于临床评估报告的撰写人资格有相应的要求。SUNGO组建了临床评估业务*技术小组,包括医学博士,国际**认证机构评审人员,世界**医疗器械企业质量经理等相关专业人员。目前SUNGO已经交付了近百种产品的临床评估报告,其中包括外科手术导航系统,骨科植入产品等较高风险和复杂程度的产品。
我公司专业办理医疗产品出口欧盟、美国、中东南美等国家的各种认证:
TUV莱茵,TUV南德,***等CE认证(MDD/MDR法规),全套CE技术文件编订, CE第四版临床评价(MEDDEV 2.7.1 Rev 4)编写,欧盟授权代表,欧盟自由销售证书,ISO13485:二016,医疗器械单一审核程序(MDSAP)审核、美国FDA注册(含FDA510K申请),FDA美国代理人服务/ FDA QSR820验厂辅导及整改,FDA警告信应对&RED LIST REMOVAL/QSR820体系辅导/OTC验厂辅导及整改,英国BRC认证咨询,BSCI验厂辅导;口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试、防护服type5/6 (PPE认证咨询(89/686/EC个人防护指令辅导)。
我公司临床报告业务优势
按照第四版临床报告指南的要求,对于临床评估报告的撰写人资格有相应的要求。SUNGO组建了临床评估业务*技术小组,包括医学博士,国际**认证机构评审人员,世界**医疗器械企业质量经理等相关专业人员。目前SUNGO已经交付了近百种产品的临床评估报告,其中包括外科手术导航系统,骨科植入产品等较高风险和复杂程度的产品。
我司将基于目前申请企业的现状,依据MEDDEV 2.7.1 Rev 4的相关要求,编制能够符合发证机构发证公司的全英文临床评估报告。
针对MEDDEV2.7.1 Rev 4,我司可以协助您:
1、协助建立临床评价程序;
2、建立临床评价方案;
3、寻找等同产品,进行等同分析;
4、搜索文献及其他临床数据;
5、临床数据分析;
6、完成临床评价报告;
7、全英文临床评估报告;
8、认证机构审核通过。
我司将基于目前申请企业的现状,依据MEDDEV 2.7.1 Rev 4的相关要求,编制能够符合发证机构发证公司的全英文临床评估报告。

医疗器械CE欧盟授权代表
为了更好地保护欧盟的消费者和环境,欧盟的法律要求,为了实现产品的可追溯性 (traceability),制造商投放到欧盟市场的加贴了CE标志的产品必须标有制造商的名称和联络地址;如果制造商来自欧洲经济区EEA(包括EU与EFTA)以外的国家,其产品必须同时标有制造商和制造商的欧盟授权代表的名称和联络地址。
为了提高整体的市场监督效率,欧盟**将负责检查市场监督效率,并要求所有成员国都应满足较低法律要求,加强合作和交流。此外,欧盟**将和海关合作,并与相关利益方(制造商、欧盟授权代表、进口商、分销商)开展合作,建立产品追溯系统。欧盟**首先关注高风险领域,比如:医疗器械。
欧盟授权代表的定义与职责
欧盟授权代表(European Authorised Representative 或European Authorized Representative)是指由位于欧洲经济区EEA(包括EU与EFTA)境外的制造商明确*的一个自然人或法人。该自然人或法人可代表EEA境外的制造商履行欧盟相关的指令和法律对该制造商所要求的特定的职责。
新方法指令要求欧盟授权代表必须位于欧洲经济区境内并且具有商业注册地址(某些国家还要求欧盟授权代表必须有公司注册号或欧盟增值税VAT注册号);
EEA成员国的**及主管机关可以随时直接找上欧盟授权代表核查EEA境外的制造商是否履行了欧盟相关的指令和法律所要求的职责;
制造商的一般商务代表(例如授权经销商),不论是否位于欧洲经济区境内,都不应该与新方法指令所要求的欧盟授权代表混淆;
虽然欧盟授权代表可代表EEA境外的制造商履行欧盟相关的指令和法律对该制造商所要求的特定的职责,但制造商依然是承担主要责任的一方。
在没有制造商同意的情况下,欧盟授权代表不可擅自单独更改EEA境外的制造商所制造的产品,即使是为了使违规产品符合欧盟产品指令的要求。
中文译名用法
欧盟授权代表在欧盟的产品指令(Directive)英文版里使用的标准术语为(英国式英语) European Authorised Representative, 因为需要*欧盟授权代表的多数为位于欧盟境外的国家的制造商,尤其以美国的制造商为多, 美国的制造商更喜欢将欧盟授权代表以美式英语书写为: European Authorized Representative。
鉴于中文并非欧盟的官方语言,因此European Authorised Representative 并没有对应的中文的官方术语。在中文的翻译里,通常将European Authorised Representative 或European Authorized Representative译为: 欧盟授权代表。 也有翻译为: 欧盟授权代理、欧洲授权代表、欧洲授权代理等。通常简称为:欧盟代表或欧代。也有使用:欧盟代理、欧洲代表、欧洲代理等。在闽台,还有使用欧体授权代表或欧体代表,尽管欧洲共同体(EC-European Community)早在1993年已被欧洲联盟(EU-European Union)取代。
为什么您需要欧盟授权代表?
为了确保欧盟境内市场上销售的产品的*性,以及确保从欧盟境外进口的加贴CE标志的产品满足欧盟对消费者、财产、和环环境保护等方面的法律法规的要求,欧盟法律对欧盟境外的制造商提出以下要求:
1) 委任欧盟授权代表(欧盟授权代理):
欧盟境外的制造商必须委任一个设立于(established in)欧盟+EFTA共30个成员国境内的欧盟授权代表(欧盟授权代理) (Authorized Representative),专门代表欧盟境外的制造商与欧洲30国的**和机构打交道。
2) 授权代表必须印在包装上:
从欧盟境外进口的加贴CE标志的产品的包装、标签和使用说明书等上面,必须清楚地印上制造商的欧盟授权代表(欧盟授权代理)的名称、地址。
3) “技术文件”必须保存于欧盟授权代表(欧盟授权代理)处:
欧盟授权代表(欧盟授权代理)处必须保存较新的、所有加贴CE标志的产品的“技术文件”(Technical Files)。根据欧盟法律,确保能随时及时地提供给欧盟境内CE监督机关检核。 在最后一批产品投入市场之后,其技术文件应在欧盟授权代表(欧盟授权代理)处保留至少5年。
4) 建立“事故防范监督系统”:
欧盟境外的制造商必须在欧盟境内建立一套有效的“事故防范监督系统”,通过其欧盟授权代表(欧盟授权代理) 对产品的事故报告、通告、召回等等提供协助。
为什么欧盟授权代表(欧盟授权代理) Authorized Representative 不同于产品的进口商或销售商 ?
1) 欧盟授权代表(欧盟授权代理)是非欧盟厂家在欧盟+EFTA共30个成员国境内专门处理有关CE事宜的授权代表。代表厂家与欧洲的**和机构打交道。就好像你要在法庭里面需要一个律师一样。欧盟授权代表(欧盟授权代理)通常不会涉及一般应由进口商或销售商做的有关产品的销售业务。许多进口商或销售商擅长于市场和营销,但未必熟悉或愿意涉及复杂的CE及法律问题。
2) 产品的制造商必须要把欧盟授权代表(欧盟授权代理)的名称、地址等信息印刷在销售到欧盟市场的产品的包装或标签上面,以便欧洲30国的**和机构能直接与欧盟授权代表(欧盟授权代理)联系。
3) 根据欧盟法律要求: 一个非欧盟厂家在欧洲30国境内只能有一个欧盟授权代表(欧盟授权代理), 但却可以有许多分销商或销售代理。 如果产品在欧洲30国市场出现问题,所有欧洲30国的**和机构直接与欧盟授权代表(欧盟授权代理)联系。因而不可把欧盟授权代表(欧盟授权代理)和众多的销售商混为一谈 。
4) 销售商的主要工作重心是在产品的销售或市场上面,而不会是在处理您的CE事务上面。而我们及时追踪较新的欧盟法规,为您符合欧盟法规保驾**。
5) 如果CE监督/主管当局对因产品的销售系统而引起的严重事故质询/询问, 我们作为您的欧盟授权代表(欧盟授权代理)将始终鉴定的维护您的利益。
6) 您愿意由不参与经销活动的、中立的伟康公司在欧盟境内保存您的技术文件呢?还是由有朝一日可能会成为你的竞争对手的、欧盟境内的销售商**来保管?
7) 如果您将来在欧洲30国境内有许多家销售商,但欧盟法律只允许您有一个欧盟授权代表(欧盟授权代理),您愿意委任其中一家为您一的欧盟授权代表(欧盟授权代理),而得罪所有其它的许多家销售商吗?
8) 为了长远利益,绝大部分多数厂家都愿意选择专业的、不参与经销活动的、中立的欧盟授权代表(欧盟授权代理)公司。
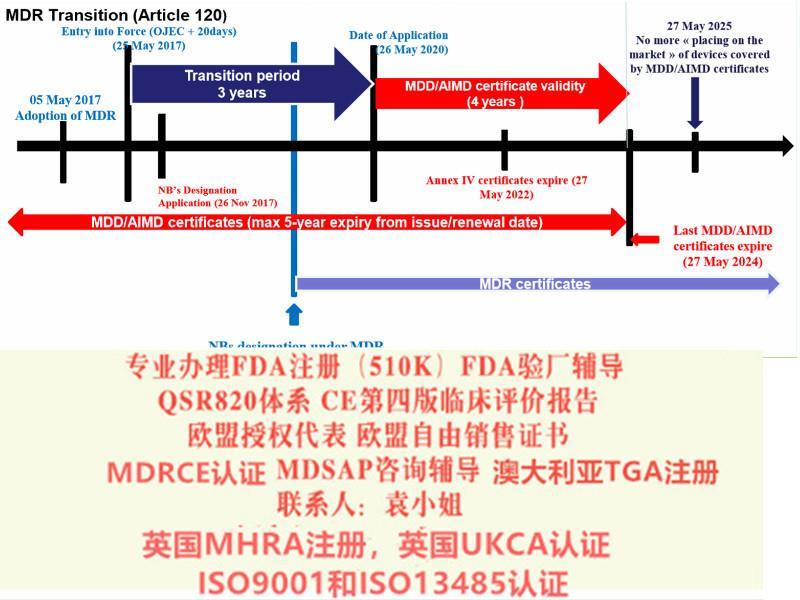
出口:美国FDA注册(含FDA510K申请)、 FDA QSR820验厂及整改、FDA警告信应对&RED LIST REMOVAL/QSR820体系/OTC验厂及整改、CE认证(CE整套技术文件编订、 CE第四版临床评价(MEDDEV 2.7.1 Rev 4)编写)、ISO13485:2016、欧盟授权代表、欧盟销售证书、英国BRC认证,BSCI验厂;口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试、防护服type5/6 (PPE认证(89/686/EC个人防护指令)。
进口商、经销商的法律责任
进口商:
应确认生产商已*欧盟代表、已签发符合性声明、已使用UDI、标签说明书符合要求、已正确使用CE标志;
- 器械或其包装或其随机文件上,应注明进口商名称、注册地址、联系地址和注册商标等信息;
- 应确认器械已在欧盟数据库注册登记,进口商自身也应登记获得SRN;
- 应确保器械在符合生产商要求的环境里存储和运输;
- 应对客户投诉、召回、不合格品记录在案,并通知生产商及欧盟代表;
经销商:
- 应确认产品上已正确使用CE标志、制造商已经签署符合性声明、标签说明书语言符合法规要求、已正确使用UDI;
- 应确认产品包装或其随机文件上有进口商的信息;
- 应确保器械在符合生产商要求的环境里存储和运输;
- 应对客户投诉、召回、不合格品记录在案,并通知生产商及欧盟代表;
- 认为产品不符合MDR&IVDR要求的,应立即通知制造商或欧盟代表或进口商;如果涉及严重风险的,应立即通知主管当局。
UDI-器械识别码
UDI是Unique Device Identification的缩写,是显示在产品标签上的一组条形码或二维码,由器械识别码DI和生产识别码PI组成,前者包括制造商和产品名称信息,后者包括产品批、有效期等信息;
制造商发货、进口商验收、销售商时都需要扫描UDI,
数据在UDI数据库中保存,从而实现产品全程可追溯;
UDI中的Basic UDI-DI也将出现在
制造商签发的CE符合性声明中,
也会显示在CE证书上;
欧盟授权GS1、HIBCC、ICCBBA
这三家美国公司管理UDI,制造
商应自行向三家机构申请产品的
UDI。
./
./
./
-/gjhdff/-
http://sungofda.cn.b2b168.com










