周期8周
费用100000
资料基本资料
机构SUNGO
产品医疗用品
器械法规(MDR)的背景
MDR将取代现有的器械指令(93/42/EEC)(MDD)和主动植入式器械指令(90/385/EEC) (AIMDD)。MDR于2017年5月发布,标志着MDD和AIMDD之间为期三年的过渡期的开始。
在过渡期间,MDR将逐步生效,首先是与公告机构和制造商根据MDR申请新证的能力有关的规定。
过渡期将于2020年5月26日,即MDR法规的“适用日期”(DoA)结束。从那时起,MDR将完全适用。
签订欧盟代表协议---起草I类器械的MDR CE技术文档---欧盟注册---
签发DOC。
这种途径符合CE的标志,就是完成如上四步,终生效的DOC是没有公
告号的。
建议企业同步建立ISO 13485器械质量体系,产品需要满足的检测
要求(比如口罩的EN14683产品性能检测;ISO 10993生物相容性检
测,还是要满足)。产品市场后的监督工作还是要有计划做。
另外,外销产品随附的文件,如说明书、标签都是要符合欧盟相关
器械标准要求EN ISO 15223 & EN 1041。
现在市面上所有发的设计精美,各式各样的所谓的发证机构发出的证
书,费用在1-2万,周期几天的,这种证,其实只是精装的DOC,如
果没有有效的欧代协议+TCF技术文档+欧盟注册作为支撑,这种证是
没有支撑证据的,也即没有真实的作用。

MDR的时间,尽快启动MDR法规合规准备事宜。
l 重新确认产品风险分类等级,确认是否有风险等级升级的情况?
例如部分可重复使用的器械,原属于ClassⅠ的器械,按照新法规变成了ClassⅠ*类器械。美容类产品原MDD下不属于范围,现MDR法规中已纳入;
l 确认原CE证的发证机构是否已获得欧盟当局批准的颁发MDR证的,目前拥有该的认证机构:BSI、TUV南德(注意TUV莱茵目前还未获得批准);

1.欧盟(非无菌):医用口罩、面罩、眼罩、防护服、隔离服和手套等非灭菌I类产品与检测试剂盒产品,提供技术文件、欧盟授权代表和注册凭证证办理服务。
备注:欧盟对于I类的产品,要求企业自己必须确保自己的产品符合欧盟法规,产品是*有效的,并建立相关技术文件,签署符合性声明,确定欧盟授权代表,然后到成员国主管当局注册登记之后,即可在产品加贴CE标志进入欧盟销售。
不需要公告机构参与审核发证的
欧盟会规定了MDR 的转换期的要求
2017 年5 月25 日:MDR 和IVDR 生效
2020 年3 月25 日:启动欧盟器械数据库(Eudamed)
2021 年5 月25 日:MDR 实施开始
2022 年5 月25 日:IVDR 实施开始
2024 年5 月25 日:AIMD,MDD 和IVDD 证将失效
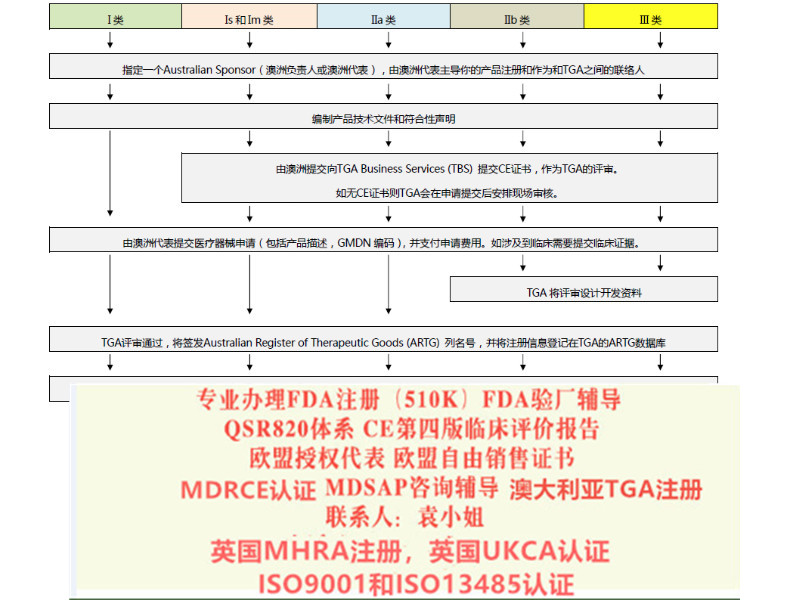
按产品的危险程度,将产品分为Ⅰ类、Is(灭菌),Im(测量),Ⅱa类、Ⅱb类、Ⅲ类
对于目前MDD和MDR的各个风险等级的产品实施时间:
1. 对于I类的产品,欧盟强制要求2021年5月25号强制实施MDR
2. 对于Is(灭菌),Im(测量),Ⅱa类、Ⅱb类、Ⅲ类迟可以用到2024年5月25号
3.对于可重复使用的产品,在MDD里属于I类,但是在MDR里属于Ir类,那么如果企业在2021年5月25号之前满足了MDD的要求,迟可以用到2024年5月25号
SUNGO可以在过渡期内为提供新旧法规的咨询服务,包括:
协助判定产品分类
协助选择合理的符合性途径
协助选择合适的认证机构
制定认证的解决方案
协助完成评估
编写CE技术文件
欧盟代表服务
http://sungofda.cn.b2b168.com