产地上海
可售地**
品牌SUNGO
型号SUNGO BV
包装纸质
建立新法规实施过渡计划 5,技术文件编制 编制符合MDR要求的技术文件(TD) 编制评估报告、生物学评价报告和风险管理等技术文件等 产品设计开发流程,确保输入及输出的完整性 确认标签、上市后监督、上市后性能跟踪方案 技术文件整改(风险管理报告,性能评估报告,GSPR等) 6,QMS建立:更新现有体系中IVDR用于QMS的要求 定制企业合规QMS系统 执行体系实施计划确保覆盖各个方面及各方面责任 7, 可追溯性UDI 建立可追溯性QMS要求 建立UDI系统程序及制度 确认UDI的规划及实施
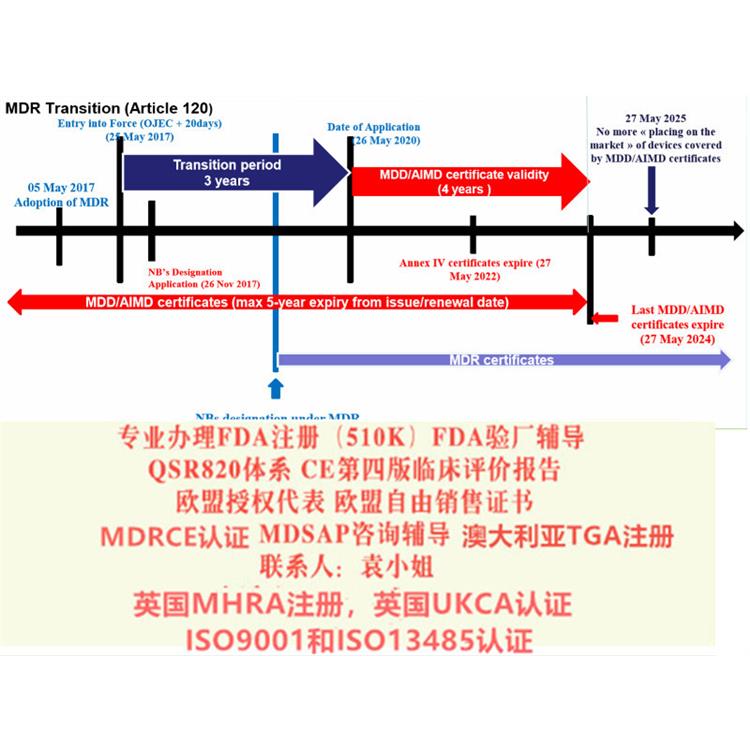
给大家带来解读MDR系列讨论 部分:演变过程和MDR的过渡期。众所周知,现行的MDD 器械指令93/42/EEC是1993颁布的,距今已经有26年的历史,这期间,器械行业无论是从技术方面,应用方面都有了巨大的变革,无疑,一部用了26年的指令已经过于陈旧,新法规替代老法规已经势在必行。2010年发生的法国PIP事件也促使了欧盟推行欧盟新法规的起草和推行。
制造商的义务经销商,制造商,进口商等的义务及其关系现已在该条例中明确规定。第10条制造商应有风险管理制度(第2款)和质量管理制度(第9款);进行评估(第3段);编写技术文件(第4段);,并采用合格评定程序(第6段)。制造商亦须对其产品在市场上销售后负责(第12、13、14段)。它们必须有适当的制度来弥补它们对有缺陷的装置造成的损害所负的财务责任(第16段)。每个制造商应一名负责合规的人员(第15条)。一些可植入设备的制造商必须为患者提供植入卡(第18条)。一旦制造商完成所有这些义务,他们应制定一份符合性声明(第19条),并在其设备上应用CE标记(第20条)。欧盟/欧洲经济区以外的制造商应与欧盟/欧洲经济区内的授权代表签订合同(第11条)。授权代表(第11条)、进口商(第13条)和分销商(第14条)的义务也作了明确说明。公告机构NB根据MDR新规定,必须公告机构。器械将被要求满足更严格的标准,特别是在能力方面。

MDD指令和MDR法规的CE认证的区别
1:老MDD指令申请CE认证,由于法规规定产品在市场上出现任何问题,都是由制造商承担。其中欧盟授权代表的职责只是沟通协调以及产品包装可以使用欧盟授权代表的公司名字和地址信息的责任。国外的进口商更多的是找工厂要一张MDD的CE证书,能顺利清关销售便可以了,一般不关注你们这个证书怎么获得的,是否正真满足法规要求的。
2:但是新MDR的管控趋于严格,对于制造商,欧盟授权代表以及国外进口商三方该承担的责任比较明确,欧盟授权代表和进口商与制造商一样为缺陷器械承担连带的法律责任。所以进口商在采购工厂产品的时候,较MDD老法规,他们更关注,工厂是否真正满足CE法规要求,尽量的将自己要承担的风险降低到。
3:我们为企业编写的MDR CE技术文件里的:风险分析报告,评价报告,基本基本检查表等等,不仅仅是为了获得一张证书而做的,更多的都是从各个方面来产品是的有效的。
需要办理以下认证可以随时找我 :
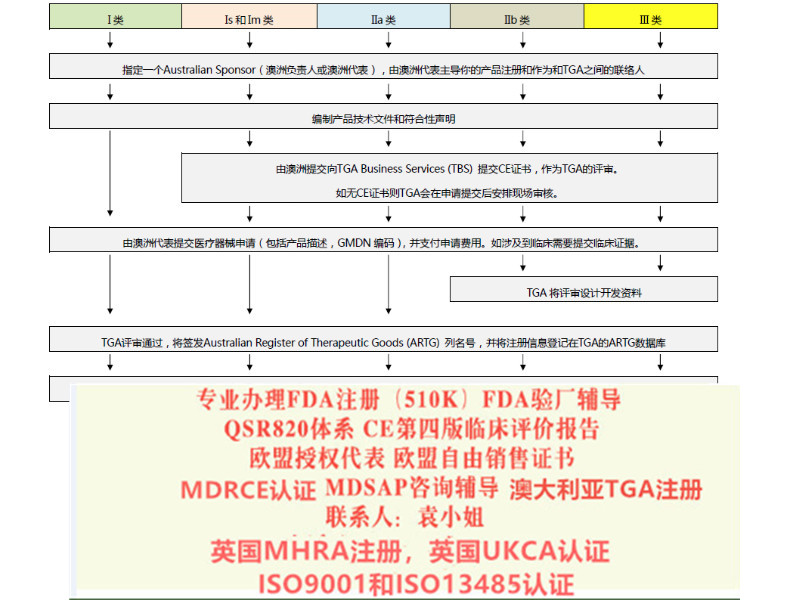
1:出口欧盟:MDR CE认证/IVDRCE认证,欧盟授权代表,欧盟注册,欧盟自由销售证书
2:出口英国:英国代表,英国MHRA注册,UKCA认证,英国自由销售证书
3:出口美国:美国FDA注册,FDA510K,QSR820体系
4:中国:国内的器械注册证和生产许可证
5:出口加拿大:加拿大的MDEL注册
6:质量管理体系认证:ISO13485咨询和认证

SUNGO公司介绍
SUNGO创建于2006年,立志于成为**化的器械法规技术服务商。目前SUNGO在中国、欧洲、北美和澳洲均设有服务机构,服务过的客户更是覆盖了(中国、越南、马来西亚、孟加拉、新加坡)、欧洲(英国、瑞士、瑞典、丹麦、挪威)、北美(美国、加拿大)、南美(阿根廷)、大洋洲(澳大利亚)和非洲(博茨瓦纳、南非)等和地区。
SUNGO致力于为**的器械生产商和经营者提供市场准入的合规咨询以及国际注册服务。从产品生产、检测、过程管理、注册、认证、整改、上市跟踪等各环节为企业提供的技术支持,为产品合规和顺利上市保驾**。
http://sungofda.cn.b2b168.com