电动轮椅的MHRA注册 供应
发货地址:上海市金山区
产品数量:123456.00个
价格:面议
规格1
密度4
宽度2
质量5
高度3
英国负责人还必须:终止与制造商的法律关系;和将该终止通知MHRA和相关的批准机构。进口商或分销商有可能担任英国负责人。
我公司办理欧盟,美国,澳大利亚以及中东南美等等各类认证:FDA510K认证,欧盟自由销售证书,欧盟授权代表,ISO13485/ISO9001认证,欧盟CE认证(MDR(REGULATION (EU) 2019/745)),FDA注册,FDA验厂,英国授权代表,MHRA注册,美国代理人服务,澳大利亚TGA认证,CE整套技术文件编订、 CE第四版评价(MEDDEV 2.7.1 Rev 4)编写)、防护服PPE指令Type5/6认证、器械单一体系审核MDSAP认证、BSCI验厂、BRC 认证,澳大利亚TGA注册、口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试,器械产品备案登记表、器械产品注册证、生产备案登记表、生产许可证 欧盟注册(英国MHRA注册)Medicines & healthcare products regulatory agency 欧盟授权代表 EC/REP:representative in the EU 如果你是一个注册在英国的企业,你可以在英国主管机关(MHRA)进行注册。如果你也有在欧盟其他成员国的企业,你可以选择向其中的一个,而不一定要是英国的,但是你必须通知你所注册的营业场所所在国的主管当局。 如果你实施的是另一个成员国注册,而不是英国的,你应该从该成员国的主管当局寻求信息

我们可以为您提供的自主服务项目主要有: 出口欧洲法规:欧盟CE认证(CE整套技术文件编订、 CE第四版评价(MEDDEV 2.7.1 Rev 4)编写)、ISO13485:2016认证、欧盟授权代表(德国,英国和荷兰)、欧盟注册、欧盟自由销售证书、防护服PPE指令Type5/6认证 出口美国法规:器械、化妆品、食品美国FDA注册(含FDA510K申请)、美国代理人、 FDA 验厂及整改、FDA警告信应对&RED LIST REMOVAL、QSR820体系、食品FDA验厂及整改、OTC药品FDA验厂及整改 中国法规:器械产品备案登记表、器械产品注册证、生产备案登记表、生产许可证、经营许可证、ISO9001/13485认证、SFDA验厂、SFDA注册检测、企业标准编制、局自由销售证。 出口其余国际法规:器械单一体系审核MDSAP认证、澳大利亚TGA注册、BSCI验厂、ISO22716 GMPC验厂、BRC 认证,口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试 欧盟注册定义 依据欧盟法规规定,所有的MDD I类器械,客户定制器械以及IVDD的OTHER类别的器械在出口到欧盟时都需要完成欧盟注册。欧盟注册是由欧盟各个成员国的器械主管机构来实施审批的。 欧盟注册需要提供的资料 所有申请者应提供企业名称,地址等基本信息以及产品的名称和型号等信息。除此之外,还需要提品的说明书(必要时)以及公司签发的DOC(符合性声明)。 欧盟注册的种类 SUNGO可以提供英国器械企管当局MHRA的注册服务,以及荷兰器械主管当局的器械注册服务
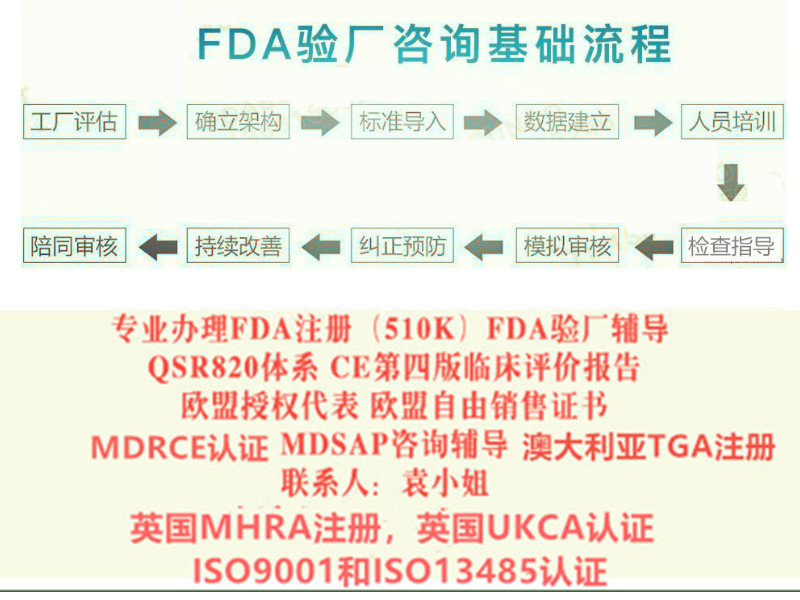
CE新版评价报告怎么编写? 实施计划 对于第四版的执行时间,各家公告机构做法有所不同, 所持的意见也不尽相同。据我们所了解,公告机构的基本思路可以简单归纳为: -高风险产品和植入器械(例如Class III 和Class IIb), 公告机构期望器械制造商马上执行。 -低风险器械(如 Class IIa, Is, Im), 公告机构会适当放宽期限,有些机构要求2019年内完成更新; 而有些机构则放宽对state of the art 的要求。 应对措施 制造商应该对第四版进行差距分析,从而: 1)对QMS(质量管理体系)的流程进行影响分析; 2)对CER(现有评价报告)进行差距分析; 3)对实际更新准备过渡计划(过渡计划应考虑与产品相关风险及证书到期日)。 如何更新CER -上市后监督信息(PMS & PMCF) -当前技术水平 (State of the art) 重要信息 PMCF 是强制的,这也是新欧盟法规MDR **的重要内容之一。对于下列情形,器械生产商需要做好充分准备: 1)之前的CER (上市评价) 走的是等同性路径(特别是高风险产品,如:Class III 和植入器械) 2)产品使用的风险高 3)针对高风险的解剖部位/ 高风险的人群 4)出现了有关性和有效性方面的新的信息 5)创新的器械 6)器械的设计适应症和预期用途发生重大的变化。 制造商应该对第四版进行差距分析,从而: 1)对QMS(质量管理体系)的流程进行影响分析; 2)对CER(现有评价报告)进行差距分析; 3)对实际更新准备过渡计划(过渡计划应考虑与产品相关风险及证书到期日)。 如何更新CER -上市后监督信息(PMS & PMCF) -当前技术水平 (State of the art) CE新版评价报告怎么做? 我司报告业务优势: 按照第四版报告指南的要求,对于评估报告的撰写人有相应的要求。我司组建了评估业务技术小组,包括博士,国际认证机构评审人员,世界**器械企业质量经理等相关人员。目前我司已经交付了近百种产品的评估报告,其中包括手术导航系统,植入产品等较高风险和复杂程度的产品。例如:一次性无菌器带针、一次性无菌输液器带针、电子体温计、润滑剂、活组织检查针、输液泵、雾化器、一次性麻醉穿刺针等产品,TUV南德/TUV莱茵/BSI/KIWA等公告机构要求的欧盟第四版器械评价/评估报告,提供编写或更新。 我司将基于目前申请企业的现状,依据MEDDEV 2.7.1 Rev 4的相关要求,编制能够符合发证机构发证公司的全英文评估报告。
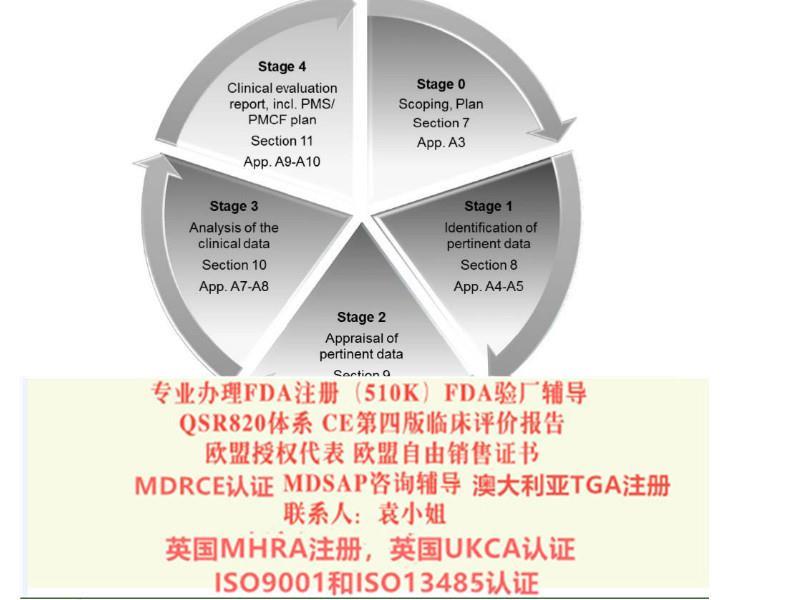
MHRA注册流程:编订相关MHRA申请文件;企业向MHRA提交注册申请
我们的核心资源包括分布在**主要经济体的运营网络,具有美国IAS认可资质的实验室,具有ANAB认可资质的认证机构,以及分布**的资源。依靠这些资源,我们为**的器械生产商和经营者提品全生命周期的市场准入服务。
http://sungofda.cn.b2b168.com










