产地上海
可售地**
品牌SUNGO
型号SUNGO BV
包装纸质
新的标识识别系统(UDI系统)(第27条)将有力地增强市场后相关活动的可追溯性和有效性。
MDR还将提高透明度,公开有关设备和研究的信息。
MDR的主要变化 1.扩大了应用范围 2.提出了新的概念和器械的定义 3.细化了器械的分类 4.完善了器械的通用和性能要求 5.加强对技术文件的要求 6.加强器械上市后的 7.完善评价相关要求 8.提出Eudamed数据库的建立和使用 9.提出器械的可追溯性(UDI) 10.对NB提出严格的要求在新版MDR 2017/745/EU中,更是完善了评估(包括器械售后追踪)和调查的执行、评估、报告和更新资料的相关要求。对特定III类和IIb类器械,评估报告中要考虑咨询小组的意见;对植入式和III类器械,提出考虑研究;要求评估报告按照售后追踪所取得的数据进行更新;

我们的咨询业务
1:MDR法规培训
新法规立法过程、变化及转换期
MDR覆盖的范围,包括和MDD, AIMD的修订要点及主要区别
MDR法规结构及条款清单
MDR分类规则要求
MDR对经销相关方 (Economic Operators)要求
MDR符合性审核程序
质量管理体系的全新要求,以及MDR与ISO 13485:2016的关系
通用和性能要求GSPR
MDR对技术文档TCF的要求
评价CER上市后追踪PMCF的要求
上市后监督PMS的要求
MDR中对器械性标识UDI要求;
欧盟符合性声明 (EU declaration of conformity) 要求
器械欧盟数据库(European Database on Medical Devices, EUDAMED)介绍及输入
公告机构的审核准则
充分准备以应对MDR欧盟器械法规相关的审核
Q&A
2:专项,MDR法规变化-UDI和标签
MDR法规变化-GSPR
MDR法规变化-PMS、PMCF系统
MDR法规变化-质量管理体系
MDR法规变化-技术文件
MDR法规变化-评估,调查
MDR法规变化-风险管理
可用性评价
3.欧代和注册服务SUNGO 荷兰和德国公司可以提供欧盟授权代表服务,同时提供向当地部门申报注册的服务。
4.整体MDR升级换版实施服务 包含上述1、2、3的全部内容,还包括针对公告机构审核开具的不符合的整改服务。
我们的服务流程
1 预评估:简要管理,以确保清楚了解MDR的重要性和业务影响
预评估考虑组织的挑战:管理意识,人员配备能力和可用性,预算影响
2.差异分析 评估对产品、内部资源、组织和预算的影响
检查新的分类规则(MDR I, IIa,IIb,III类),确认现有和产品的符合性评估路线
核对器械定义,确认是否属于扩大范围或属于附录16中所涉及的属于器械的范围
检查产品与关NB机构有关的要求
审查现有技术文档(技术文件)的变更
评估及更新质量管理体系(以下第3点)
检查可用证据和风险管理的充分性,识别差异(第56条)
评估产品标签(附件I第Il1章)
确保上市后监督的安排充分适宜够(第七章第1节)
制定上市后性能跟踪(PMCF,附件XIV B部分)
做好迎接新的警戒需求的规定(第七章第二节)
确保可追溯相关方面的义务(第3章)
3,质量体系评估 评估新IVDR法规下QMS符合标准和流程的程度
增加新法规应用于QMS的要求
协助和识别合规负责人(PPRC)并参与培训
4,公告机构NB确认 选择合适的公告机构,确认公告机构资质及范围
建立新法规实施过渡计划
5,技术文件编制 编制符合MDR要求的技术文件(TD)
编制评估报告、生物学评价报告和风险管理等技术文件等
产品设计开发流程,确保输入及输出的完整性
确认标签、上市后监督、上市后性能跟踪方案
技术文件整改(风险管理报告,性能评估报告,GSPR等)
6,QMS建立:更新现有体系中IVDR用于QMS的要求
定制企业合规QMS系统
执行体系实施计划确保覆盖各个方面及各方面责任
7, 可追溯性UDI 建立可追溯性QMS要求
建立UDI系统程序及制度
确认UDI的规划及实施

产品名称和商品名、产品代码、目录编号或欧盟符合性声明中包含的其他允许识别和追溯产品的明确的参考号,如适当照片,以及适当时其预期目的。除产品或商品名称外,第3条中基本UDI – DI所提供的允许识别和可追溯产品的信息
按照附录VIII的规则所划分的器械风险等级
当前声明所涵盖的器械符合本法规,以及适用时其他相关的要求签署欧盟符合性声明的欧盟立法的声明

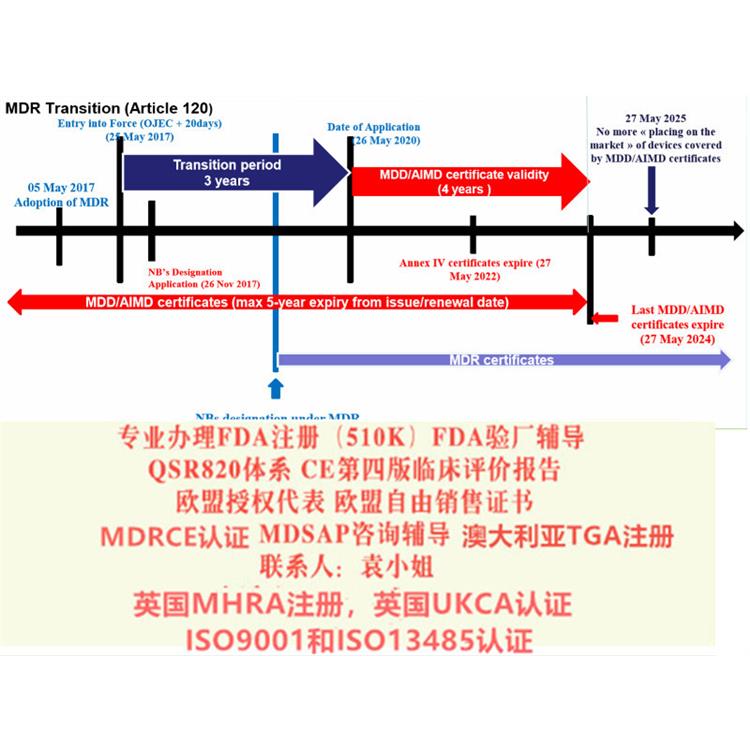
欧盟器械法规MDR (EU)2017/745第120条过渡性条款(Article 120 Transitional Provisions)应运而生。
MDD CE的持证制造商应符合Article 120条款要求,避免在宽限期严重不符合项的产生。
Article 120中需要关注的2个时间点:
1、2020年5月26日后,CE证书上的任何内容不再发生变化,已上市产品不能发生重大变更,如有变更,需要重新申请MDR证书。
2、2024年5月27日,MDD&AIMDD颁发的所有证书失效。
MDD CE的持证制造商需关注的两个方面:
1、基于MDD/AIMDD投放市场的产品,在2025年5月27日前可在市场上继续流通或投入使用。即若产品已进入市场且证书在有效期内,那么产品在市场上可以继续流通、使用,截至2025年5月27日。
2、2020年5月26日后,产品需满足MDR新规关于上市后监督、警戒、市场、经济营运商与产品注册的相关要求,以上要求对于企业至关重要,请听S君细细道来。
需要办理以下认证可以随时找我 :
1:出口欧盟:MDR CE认证/IVDRCE认证,欧盟授权代表,欧盟注册,欧盟自由销售证书
2:出口英国:英国代表,英国MHRA注册,UKCA认证,英国自由销售证书
3:出口美国:美国FDA注册,FDA510K,QSR820体系
4:中国:国内的器械注册证和生产许可证
5:出口加拿大:加拿大的MDEL注册
6:质量管理体系认证:ISO13485咨询和认证
在过渡期结束后,制造商是否仍然可以投放市场/投入使用符合指令的设备? 是的,在某些条件下,可以选择继续投放市场/投入使用符合指令的设备,直到其现有证到期为止。 这可以避免在MDR下立即需要新证。 要使用此选项,所有现有证必须有效(例如,QMS),设备的目的和性质不得更改,并且您必须遵循新的MDR规则进行注册,监视和警惕。
http://sungofda.cn.b2b168.com