周期4周
品牌SUNGO
公司SUNGO
流程SUNGO
国家欧洲
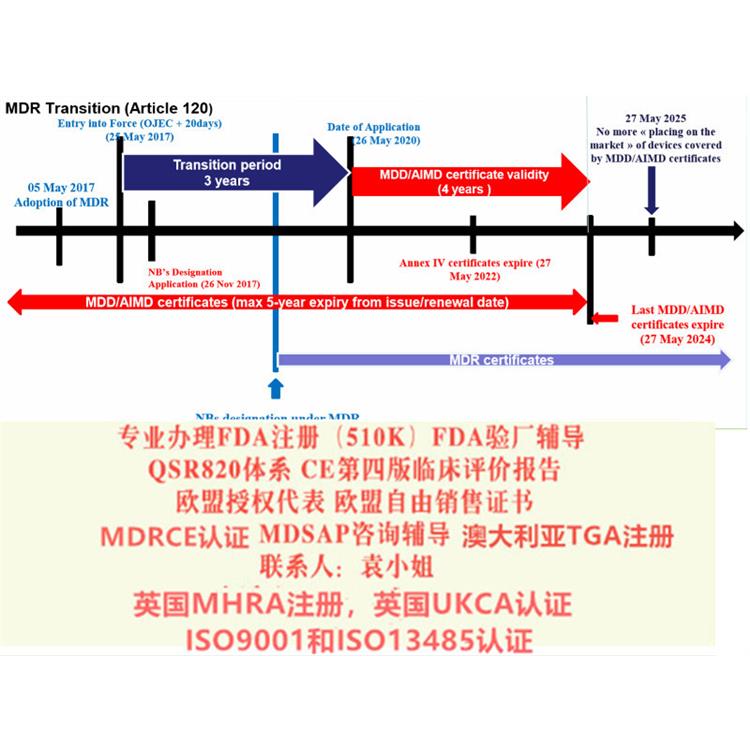
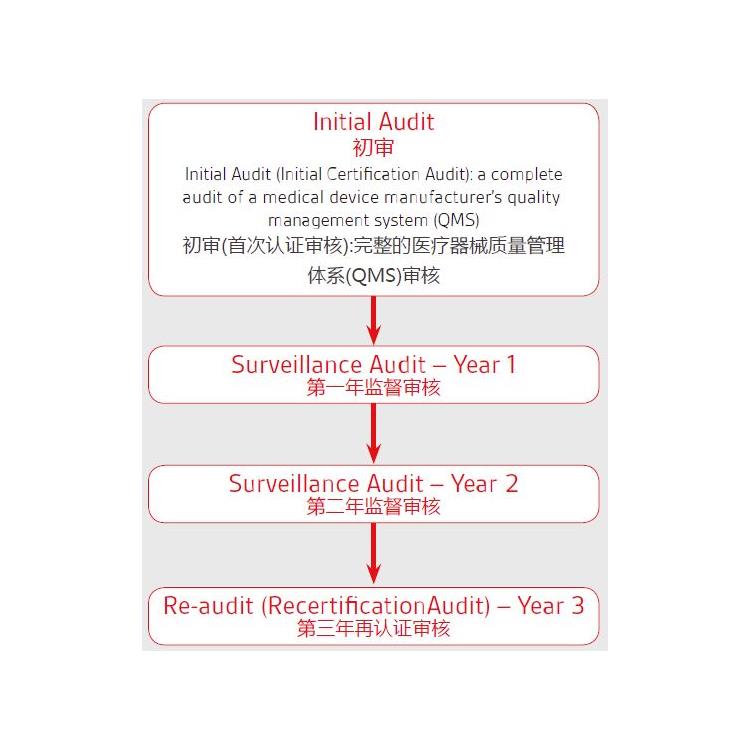
法规条款增加,认证评审更加严格,评价报告。MDR要求企业提供第四版评估报告,相比于第三版,第四版要求更为严格;
MDR 第17 条规定,一次性使用的器械的复用只能在相应法律允许的情况下进行,且应符合MDR 的规定。任何对一次性使用器械的再处理即复用的自然人或法人应视为复用器械的制造商,承担制造商义务,包括器械的可追溯性。但目前只有部分欧盟成员国接受器械复用并具备相应的法规规定。

出口美国需要的为: FDA注册,FDA510K,QSR820体系(美国FDA验厂) 出口非欧盟为: 国外的客户,想要进口中国工厂的产品,需要客户,先要把产品在当地的局进行注册,完成注册后,才可以进口,销售。

评价在器械性和有效性确认过程中扮演着非常重要的角色,评价作为CE认证必须的技术文档之一,其对于**产品的性能及是十分重要的,那么评价包含哪些内容呢?MEDDEV 2.7/1 rev4中给出了明确的说明,具体如下:
1、什么是评价
评估是一种持续收集,评估和分析有关器械的数据的过程,通过对这些数据的评估及分析来确认是否有足够的证据来确认在依据制造商的说明书使用器械时,其和性能符合相关基本要求。评估是制造商的责任,其报告作为器械技术文档的一部分。
2、为什么要进行评价
在器械上市前,通过对其进行评价,(1)评估产品是否达到了预期目的;(2)在考虑预期性能的风险/获益时,已知的、可预见的风险和不良事件是否降到、并是可接受的水平;(3)所有有效的声明是否都有足够的证据支持。
当器械上市后,需要通过建立的警戒系统和跟踪,对预期风险进行评估,以及**并发症,大规模使用下的及性能进行监测,并及时更新评估报告,为患者的生命健康提供**。
3、什么时候开始进行评价及评价更新要求?
评价是贯穿于器械整个生命周期的一个持续的过程:
(1)评价在符合性评价阶段开展,将器械的上市。
(2)随着在使用过程中有关该器械的新的性与性能信息的获得,对其进行周期性重复评估。
评价更新要求:
1.当制造商从PMS收到新的信息,有可能改变当前的评估;
2.如果没有收到新的信息:
(1)当器械带有重大风险或运行不好,则需每年更新一次;
(2)如果器械无明显风险且运行良好,需2-5年更新一次,但需要给出合理的理由。
4、怎样进行评价
评价是基于可利用的上市前和上市后数据的综合分析,包括性能数据和数据。
新版器械评价指南MEDDEV 2.7/1 revision 4,将评价分为0-4个阶段,在评价过程中,阶段往往是反复的。事实上,和分析阶段可能会发现新的信息并提出新的问题,需要扩大评估范围,完善评估计划,并检索、评价和分析额定的数据。
欧盟器械CE认证之——评价
阶段0:确定评估的范围及计划
评估开始之前,制造商应根据基本要求,需要从角度及器械历史及性质定义评估的范围及计划。
阶段1:相关数据的识别
用于评估的数据总的来说主要来自两个方面:制造商生成和持有的数据以及文献中的数据。其中,制造商生成和持有的数据包括:
(1)所有的上市前的研究;
(2)制造商在欧洲或其他进行的风险管理活动以及PMS项目;
(3)相关的前研究。
阶段2:相关数据的评估
为了确定阶段1获得的数据是否有价值,评估者应评估每一份文件对其性能和性的贡献。为了确保系统的评价数据,评估者在评估之前应当建立一个评估计划,用于确定评估过程和标准。
阶段3:数据分析
数据分析阶段的主要目标是确定当器械按其预期目的使用时,评估可供器械使用的的数据集是否能符合器械性能和的基本要求。为符合性,评估者应从以下几个方面考虑:
(1)使用合理的方法;
(2)综合分析;
(3)考虑是否有增加研究及其他措施的必要;
(4)确定PMCF的需求。
阶段4:撰写评价报告 (可提供代理编写或更新:TUV南德/TUV莱茵/***/BSI/KIWA等公告机构要求的欧盟第四版器械评价/评估报告、CE整套技术文件编订服务。)
对于所有的评价结果,将以报告的形式输出,即为评价报告。评价报告总结并汇总了所有相关的数据,并在技术文档的其他部分进行了记录或参考。
5、谁应该实施评价
评价应由合适的个人或团队进行。
制造商定义评价者的需求;
a)制造商应能够通过参考他们的和经验,来对评估者的选择,并为每一个评价者提出一个利益声明。
b)作为一般原则,评价者应具备以下知识:
●研究方法学(包括试验设计和生物统计学)
●信息管理(例如学科背景或图书馆员;相关数据库经验)
●法规需求
●写作(如有关科学或方面的研究生经验;有写作、系统复习和数据评估方面的培训和经验。
c)对于评估中的特定设备,评估者应该添加以下知识:
●器械的技术和应用
●诊断和管理的条件拟被设备、替代品的知识、标准和技术诊断或管理(例如相关的)
d)评价者应该在相关领域具有以下培训和经验:
●在相关领域接受高等教育的学位和5年的经验;或
●如果没有相关学位,需要10年的经验。

管理条例规定的职责和具体内容 1)代表制造商; 2)应要求向主管当局提供制造商授权委托其为EAR的副本; 3)验证制造商起草的欧盟符合性声明和技术文件; 4)在适用的情况下,验证制造商是否已执行适当的合格评定程序; 5)保留一份技术文件、符合性声明的副本,如果适用,还应保留一份相关证书的副本,供主管当局使用; 6)遵守注册义务; 7)验证制造商设备注册所需承担义务的符合性; 8)应要求向主管当局提供必要的信息和文件,以设备的一致性; 9)向制造商发送主管当局对样品或设备访问的任何请求,并验证主管当局是否收到样品或获得设备访问权限; 10)与主管当局合作,采取任何预防或纠正措施,以或减轻设备造成的风险; 11)向制造商通报人员、患者和用户对其设备相关疑似事件的投诉和报告; 12)应在与制造商相同的基础上对有缺陷的设备承担法律责任,并与制造商承担连带责任。 [img/new19/sungo99/1565589799.jpg[/img] (三)如何选择EAR
我公司办理:出口欧盟的新MDR和IVDR的CE,欧盟授权代表,欧盟注册,BASCI UDI申报
http://sungofda.cn.b2b168.com