周期4周
品牌SUNGO
公司SUNGO
流程SUNGO
国家欧洲
医用防护服、隔离衣、手术衣;产品在进入欧盟市场前,企业需根据产品的预期用途,结合欧盟器械法规(EU)2017/745中附录Ⅷ要求,将产品分为I类、II类及III类,产品风险等级越高,分类等级越高,进入欧盟市场要求越高。
我们的咨询业务 1:MDR法规培训 新法规立法过程、变化及转换期 MDR覆盖的范围,包括和MDD, AIMD的修订要点及主要区别 MDR法规结构及条款清单 MDR分类规则要求 MDR对经销相关方 (Economic Operators)要求 MDR符合性审核程序 质量管理体系的全新要求,以及MDR与ISO 13485:2016的关系 通用和性能要求GSPR MDR对技术文档TCF的要求 评价CER上市后追踪PMCF的要求 上市后监督PMS的要求 MDR中对器械性标识UDI要求; 欧盟符合性声明 (EU declaration of conformity) 要求 器械欧盟数据库(European Database on Medical Devices, EUDAMED)介绍及输入 公告机构的审核准则 充分准备以应对MDR欧盟器械法规相关的审核 Q&A 2:专项,MDR法规变化-UDI和标签 MDR法规变化-GSPR MDR法规变化-PMS、PMCF系统 MDR法规变化-质量管理体系 MDR法规变化-技术文件 MDR法规变化-评估,调查 MDR法规变化-风险管理 可用性评价 3.欧代和注册服务SUNGO 荷兰和德国公司可以提供欧盟授权代表服务,同时提供向当地部门申报注册的服务。 4.整体MDR升级换版实施服务 包含上述1、2、3的全部内容,还包括针对公告机构审核开具的不符合的整改服务。 我们的服务流程 1 预评估:简要管理,以确保清楚了解MDR的重要性和业务影响 预评估考虑组织的挑战:管理意识,人员配备能力和可用性,预算影响 2.差异分析 评估对产品、内部资源、组织和预算的影响 检查新的分类规则(MDR I, IIa,IIb,III类),确认现有和产品的符合性评估路线 核对器械定义,确认是否属于扩大范围或属于附录16中所涉及的属于器械的范围 检查产品与关NB机构有关的要求 审查现有技术文档(技术文件)的变更 评估及更新质量管理体系(以下第3点) 检查可用据和风险管理的充分性,识别差异(第56条) 评估产品标签(附件I第Il1章) 确保上市后监督的安排充分适宜够(第七章第1节) 制定上市后性能跟踪(PMCF,附件XIV B部分) 做好迎接新的警戒需求的规定(第七章第二节) 确保可追溯相关方面的义务(第3章) 3,质量体系评估 评估新IVDR法规下QMS符合标准和流程的程度 增加新法规应用于QMS的要求 协助和识别合规负责人(PPRC)并参与培训 4,公告机构NB确认 选择合适的公告机构,确认公告机构资质及范围 建立新法规实施过渡计划 5,技术文件编制 编制符合MDR要求的技术文件(TD) 编制评估报告、生物学评价报告和风险管理等技术文件等 产品设计开发流程,确保输入及输出的完整性 确认标签、上市后监督、上市后性能跟踪方案 技术文件整改(风险管理报告,性能评估报告,GSPR等) 6,QMS建立:更新现有体系中IVDR用于QMS的要求 定制企业合规QMS系统 执行体系实施计划确保覆盖各个方面及各方面责任 7, 可追溯性UDI 建立可追溯性QMS要求 建立UDI系统程序及制度 确认UDI的规划及实施
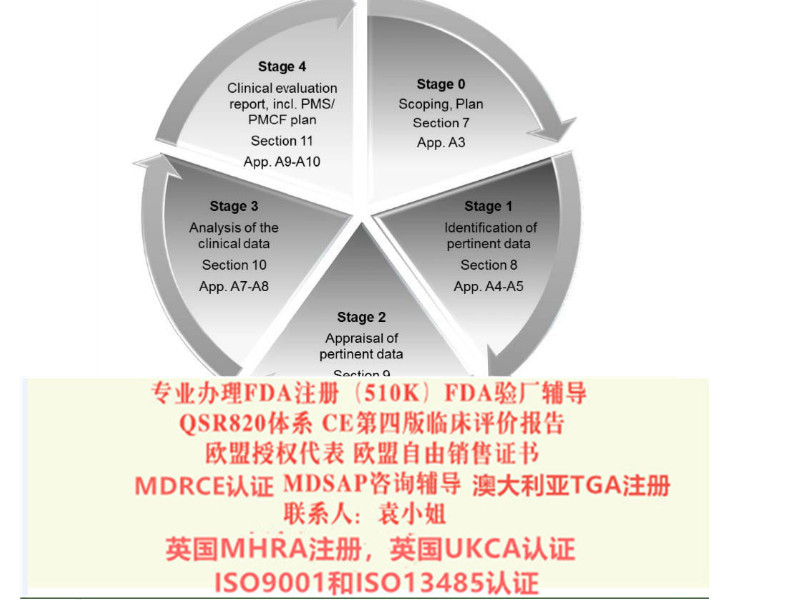
本指南为CE-器械指令应用问题相关的指南中的一部分,是关于评价资料撰写的一些原则,在MEDDEV 2.7.1 第三版的基础上增加了一部分内容,总体变更内容如下:
内容更多、更详细
提供更多有益的和案例
明确了现有要求,而非只是介绍
对于制造商应如何进行一个健全、系统的评价,以及如何数据和结论的科学有效性,有了更明确的
结合了欧盟器械法规(MDR),我们相信这将帮助器械制造商应对从指令到法规的过渡
PMS: Post Market Surveillance 上市后的监督
PMCF: Post Market Clinical evaluation 上市后的随访
4.CER明确、可衡量的目的
关于MEDDEV2.7.1 Rev 4,可以协助您:
1、协助建立评价程序;
2、建立评价方案
3、寻找等同产品,进行等同分析;
4、搜索文献及其他数据;
5、数据分析;
6、完成评价报告。

对于需要公告机构介入的器械,符合性声明的签署通常是符合性评定程序的一步。在未取得公告机构签发的CE书之前,制造商无法签署正式的DOC文件。不过制造商可以先起草一份DOC的草案提供给公告机构审查。而对于*公告机构介入的器械,制造商在法规所要求的产品符合基本要求的据准备充分后,即可签署DOC。 DOC作为法规要求的重要文件,制造商应该按照质量管理体系中文件控制程序的要求进行管控。DOC中任何内容发生了变更,则需重新签发。特别需要注意的是,对于由公告机构发的产品,DOC中任何内容的变更,都需得到原发公告机构的评审和批准。
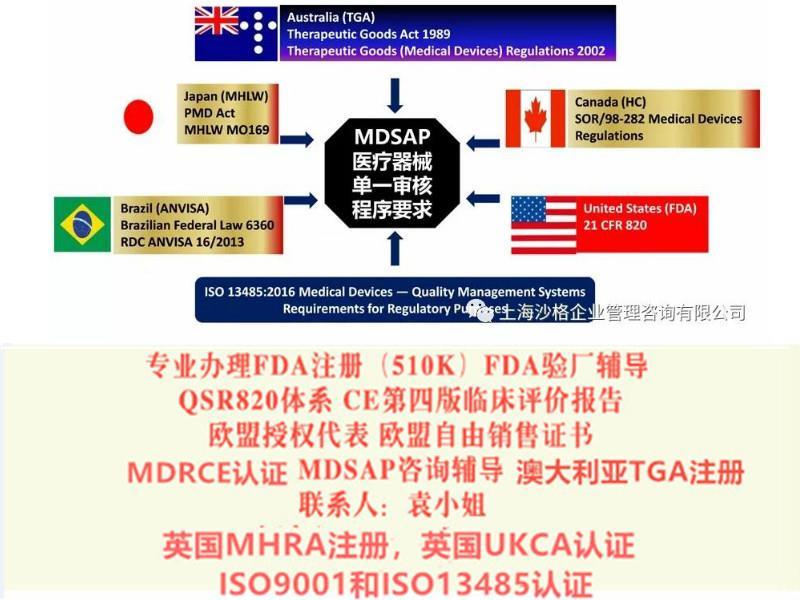
我公司办理产品出口欧盟、美国、中东南美等的各种认:
TUV莱茵,TUV南德,***等CE认,CE技术文件编订, CE第四版评价(MEDDEV 2.7.1 Rev 4)编写,欧盟授权代表,欧盟自由销售书,ISO13485:二016,美国FDA注册(含FDA510K申请), FDA QSR820验厂及整改,FDA警告信应对&RED LIST REMOVAL/QSR820体系/OTC验厂及整改,英国BRC认咨询,BSCI验厂;口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试、防护服type5/6 (PPE认咨询(89/686/EC个人防护指令)。
关于CE认的技术文件里第4版评价(MEDDEV 2.7.1 Rev 4)的背景:MED 2_7_1 Rev 4的发布对评估报告提出了新的要求,所有打算申请CE认或者已经取得CE认需要继续维持书有效的所有企业,都需要按照新版指南更新评估报告。
新版指南的变化: 相对于之前的报告,第四版的报告主要变化体系在:1.报告更新的频率,2.报告编写人和评价人的资质,3.评估报告需要有明确的可测量目标,4.确定技术发展水平,5.数据的科学性和有效性,6.比对器械,7.比对器械的数据获得,8.什么时候需要试验,9.售后监督和售后跟踪,10.风险—收益。
我公司报告业务优势:按照第四版报告指南的要求,对于评估报告的撰写人有相应的要求。SUNGO组建了评估业务技术小组,包括博士,国际认机构评审人员,世界**器械企业质量经理等相关人员。目前SUNGO已经交付了近百种产品的评估报告,其中包括手术导航系统,植入产品等较高风险和复杂程度的产品。
我司将基于目前申请企业的现状,依据MEDDEV 2.7.1 Rev 4的相关要求,编制能够符合发机构发公司的全英文评估报告。
针对MEDDEV2.7.1 Rev 4,我司可以协助您:1、协助建立评价程序;2、建立评价方案;3、寻找等同产品,进行等同分析;4、搜索文献及其他数据;5、数据分析;6、完成评价报告; 7、全英文评估报告;8、认机构审核通过。
CE技术文件编订,器械出口企业在申请CE认时,不管是I类普通产品还是II/III类高风险产品,都必须要提供第四版报告。CE技术文件编订已经拿到CE书的企业,今年监督审核也需要提供。CE技术文件编订在TUV,***等公告机构监督审核报告中明确开出不符合项目要求该版本的要求针对于MDD指令和AIMD指令,CE技术文件编订,将要申请或者已经拿到了TUV莱茵、TUV南德、***或其他公告机构CE书的企业,一定要高度关注。
我公司办理:2022年已经获得30多个K号(产品主要有电动轮椅/手动轮椅/电动代步车/口罩/手术衣/隔离衣/手套等等)
http://sungofda.cn.b2b168.com











