周期4周
包装纸质
品牌SUNGO
是否进口否
可售卖地**
sungo可以办理ISO13485咨询和认证
加拿大MDEL注册
根据加拿大器械法规(CMDR),按产品风险程度将器械分为 I, II, III和 IV四个等级, 风险等级逐次递增,I类器械为风险,IV类器械风险为。此针对制造者提出的产品注册要求也是逐级增加,要求制造者实行的体系是愈加详尽。
如果您计划进入加拿大市场,则需要进行产品注册登记以获得许可证,加拿大颁发器械机构许可证(MDEL)和器械许可证(MDL)两种不同类型证,并有不同要求
加拿大器械机构许可证(MDEL)
1. 什么是MDEL
MDEL是Medical Device Establishment License 的简称,即器械机构许可证。如果您准备在加拿大生产、进口或分销I类器械,您必须获得加拿大器械机构许可证(MDEL)。MDEL是对于机构的经营许可,无论器械属于哪个类别,器械经销商与进口商都必须获得MDEL许可证。
2. MDEL申请基本流程
(1) 企业为申请Medical Device Establishment License (MDEL)准备相应的文件,
(2) 提交MDEL申请,支付行政收费。
(3) 申请评审通过,将在Health Canada网站公示。
3. 下列几类情况能够豁免 :
(1)零售
(2)企业
(3)在加拿大市场销售企业名下获得器械产品注册证的II类III类和IV类的器械产品的制造厂商 (注:如果是市场销售其他企业名下获得器械产品注册证的II类III类和IV类的器械产品的制造厂商 ,也必须申请办理MDEL )
(4)I类器械产品的制造厂商,根据拥有器械产品营业许可证(MDEL)的企业在加拿大市场销售器械产品 ,则*申请办理MDEL
4. MDEL的有效期限
MDEL没有标明有效期限 ,但MDEL持有者一定要在每年4月1日前递交年度审核申请,MDEL才被视为继续有效,否则MDEL会被撤销 。
MDEL被撤销后,在加拿大的器械市场销售活动则也会被禁止。如果MDEL被撤销 ,MDEL持有者一定要再次申请办理并缴费,才能够再次获得MDEL,申请成功后,会发放一个新的MDEL许可证。
5. 申请MDEL需要完成申请表的如下内容,提交后2-4周可以获得证。
(1) 公司名称及联系方式
(2) 许可文件、邮寄和帐单
(3) 分类和活动表
(4) 场地
(5) 制造商信息
(6)
(7) 签名

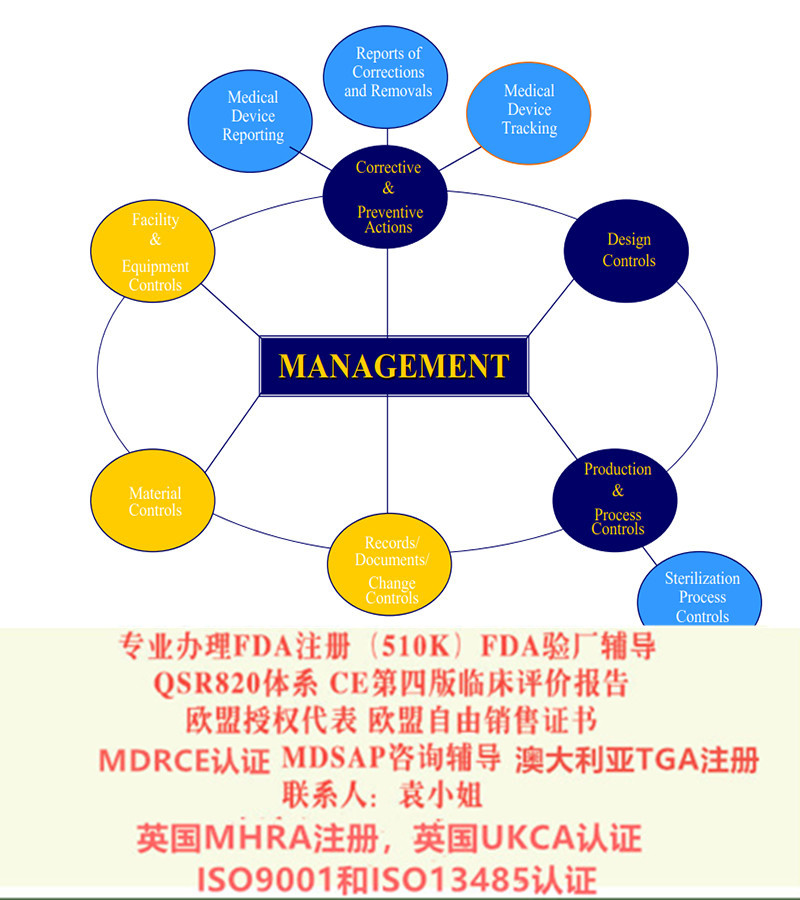
CE技术文件或设计文档(Class III)是相当于国内(中国)在产品上市前递交给局进行审评的注册文件,是对所涉及器械的一个综合*的的描述,旨在表明产品符合欧洲指令的要求。因此,编写产品
的技术文件或设计档案是欧洲CE认证过程中的非常关键的步骤,特别是在目前欧洲和公告机构对技术文档的和评估日益加严的背景下,制造商准备的CE技术文件的质量往往成为CE认证的核心和瓶颈。
技术文件或设计文档包括有关器械的设计、功能、组成、制造过程、使用、声称和评估的详细信息。它们是所有类别的设备(I类,I类无菌,I类测量,IIa,IIb和III类)所必需的,但是由于器械的种类和涉及的制造、评估过程的差异,没有两个文件是相同的。
根据多年的法规实践,峦灵建议将技术文件分为两部分:
A部分
(概要)
制造商信息:名称、、生产场地
产品名称、分类
公告机构的信息以及合格评估路径
符合性声明
产品基本介绍:预期用途、规格型号、附件等
标签、使用说明书及语言的要求
基本要求检查表
风险分析和控制的概述
产品符合的法规和标准
产品验证和确认的概述
评价报告
B部分 其余技术相关内容,如:
产品详细信息
基本要求的支持性证据
测试报告
数据
风险管理文档
过程确认
制造、检验的文件
应用的标

我们该怎么办?
l 重新确认产品风险分类等级,确认是否有风险等级升级的情况?
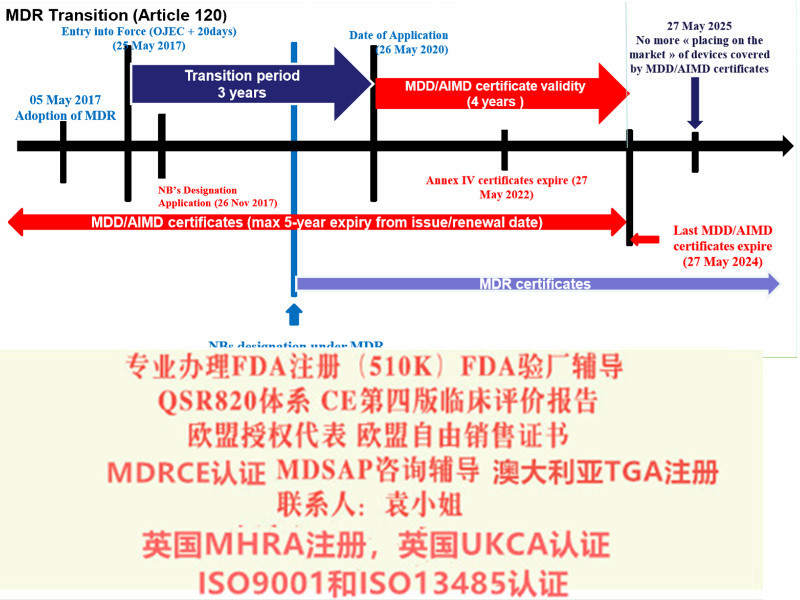
例如部分可重复使用的器械,原属于ClassⅠ的器械,按照新法规变成了ClassⅠ*类器械。美容类产品原MDD下不属于范围,现MDR法规中已纳入;
l 确认原CE证的发证机构是否已获得欧盟当局批准的颁发MDR证的,目前拥有该的认证机构:BSI、TUV南德(注意TUV莱茵目前还未获得批准);
l 确认原CE认证时的技术文件中是否含有按照Rev4原则提供的评价报告;
l 确定企业合规负责人(MDR法规要求),有相应能力、和经验来承担相应的法规工作职责。
l 修改原CE技术文件,建立质量管理体系,向具有MDR发证的认证机构提出MDR-CE认证申请,获得MDR法规下的新CE证。
目前急需做CE认证的客户很多,检测认证公司,代理公司也是鱼目混
杂,参差不齐。
怎么辨别口罩CE认证的真假呢?
1,凡是说3、5两千可以出CE证的都是
2,凡是说3/2天可以*的都是
3,凡是说国内公司自己可以发CE证的都是
辨别证的真伪很重要,因为一旦货到了港口清不了关损失就大了。
所以不要贪图*和快而上当受
NB号的CE证。按照现行的欧盟器械法规,此路径获取CE证的
时间至少8个月以上,费用比较昂贵。
欧盟I类器械,如果是非灭菌产品,具体分类为I类。按照指令要求,
公告机构不可强制介入I类器械的发证事宜,制造商可以基于完整的CE技
术文档宣称符合CE(自我CE符合性声明格式如下)。也即,制造商不需
要找发证机构去申请带NB号的CE证,制造商根据自己出具自我CE符
合性声即可打上CE mark,出口欧盟。在欧盟有经销活动前,需要委托
欧盟授权代表去当地主管机构做欧盟注册,整个欧盟28个成员国,只需
登记一次。整个周期是1-4周。
---签订欧盟代表协议---起草I类器械的MDR CE技术文档---欧盟注册---
签发DOC。
这种途径符合CE的标志,就是完成如上四步,终生效的DOC是没有公
告号的。
建议企业同步建立ISO 13485器械质量体系,产品需要满足的检测
要求(比如口罩的EN14683产品性能检测;ISO 10993生物相容性检
测,还是要满足)。产品市场后的监督工作还是要有计划做。
另外,外销产品随附的文件,如说明书、标签都是要符合欧盟相关
器械标准要求EN ISO 15223 & EN 1041。
现在市面上所有发的设计精美,各式各样的所谓的发证机构发出的证
书,费用在1-2万,周期几天的,这种证,其实只是精装的DOC,如
果没有有效的欧代协议+TCF技术文档+欧盟注册作为支撑,这种证是
没有支撑证据的,也即没有真实的作用。
SUNGO所有客户都有一对一的客服对接以保持经常性的联系,提供在线即时服务,针对贸易中存在的任何技术壁垒方面的问题提供的支持和解。
http://sungofda.cn.b2b168.com