周期4周
包装纸质
品牌SUNGO
是否进口否
可售卖地**
SUNGO从产品生产、检测、过程管理、注册、认证、整改、上市跟踪等各环节为企业提供的技术支持,为产品合规和顺利上市保驾**。
Class III,IV:
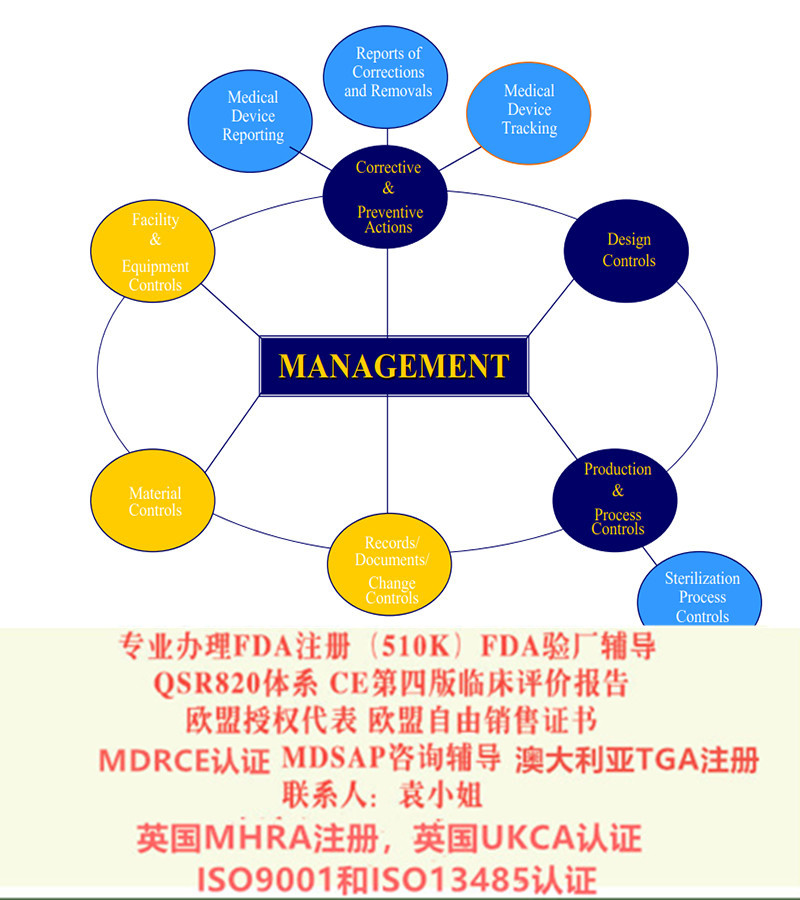
1. 通过CMDCAS认可的认证机构进行ISO 13485 审核认证(体系审核除ISO13485要求外还要包括CMDR的要求),获得证。
2. 准备Canadian Medical Device License (MDL)申请。
3. 提交MDL申请和Premarket review documents,并交纳行政收费。
4. Health Canada评审MDL申请和Premarket review documents, 评审通过后进行网站公示。
需要准备的文件清单
I类器械豁免注册。II,III,IV类器械的注册要求如下:
1. 通用注册资料:
a) 器械的名称;
b) 器械的分类
c) 器械的标识;
d) 产品标签上出现的制造者名称、;
e) 若制造地点与
f) 不同,则制造地名称、;
2. II 类器械注册附加资料:
a) 所制造、销售或代理的器械关于条件的目的及用途的描述;
b) 为满足*和有效性要求所符合的标准的清单;
c) 由制造者的高层主管作的*有效性符合声明;
d) 由制造者的高层主管作的器械标签符合加拿大器械法规的声明;
e) 若是近病人体外诊断设备(即不在而是在例如家庭使用的设备),制造者的高层主管应声明已用代表预期使用者的人体物质在与预期使用条件类似的条件下进行了研究性测试;

CE技术文件或设计文档(Class III)是相当于国内(中国)在产品上市前递交给局进行审评的注册文件,是对所涉及器械的一个综合*的的描述,旨在表明产品符合欧洲指令的要求。因此,编写产品
的技术文件或设计档案是欧洲CE认证过程中的非常关键的步骤,特别是在目前欧洲和公告机构对技术文档的和评估日益加严的背景下,制造商准备的CE技术文件的质量往往成为CE认证的核心和瓶颈。
技术文件或设计文档包括有关器械的设计、功能、组成、制造过程、使用、声称和评估的详细信息。它们是所有类别的设备(I类,I类无菌,I类测量,IIa,IIb和III类)所必需的,但是由于器械的种类和涉及的制造、评估过程的差异,没有两个文件是相同的。
根据多年的法规实践,峦灵建议将技术文件分为两部分:
A部分
(概要)
制造商信息:名称、、生产场地
产品名称、分类
公告机构的信息以及合格评估路径
符合性声明
产品基本介绍:预期用途、规格型号、附件等
标签、使用说明书及语言的要求
基本要求检查表
风险分析和控制的概述
产品符合的法规和标准
产品验证和确认的概述
评价报告
B部分 其余技术相关内容,如:
产品详细信息
基本要求的支持性证据
测试报告
数据
风险管理文档
过程确认
制造、检验的文件
应用的标
欧盟授权代表
对于在欧盟市场流通的产品,为了实现产品的可追溯性以及便于和保护欧盟消费者等目的,欧盟在某些特定指令中要求欧洲经济区以外国家的制造商必须欧盟授权代表(European Authorised Representative,简称EAR)履行欧盟相关的指令和法律对该制造商所要求的特定的职责。简言之,欧盟对高风险领域(器械)实施了设立欧盟授权代表要求,是便于直接,落实责任而制定的法律要求。

CE技术文件
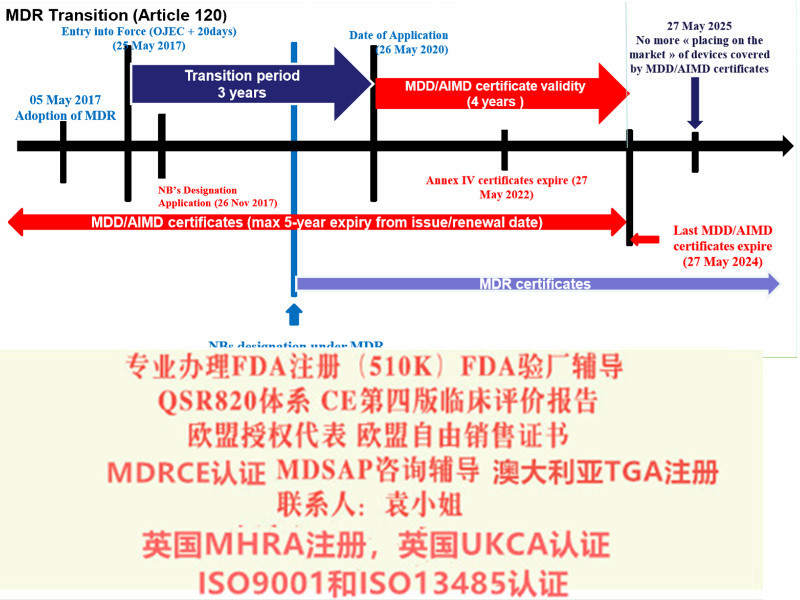
2017年2月Regulation (EU) 2017/745 on Medical Devices器械法规(MDR)提案发布,同年3月,欧盟成员国一致表决同意MDR。2017年5月5日,欧盟Official Journal正式对外宣MDR法规内容。MDR新法规将取代现行的有源器械指令Council Directive 90/385/EEC on Active Implantable Medical Devices (AIMDD) (1990)以及器械Council Directive 93/42/EEC on Medical Devices (MDD) (1993)指令。原计划2020年5月26日正式实施的MDR受**影响将推迟实施时间至2021年5月26日。
在此期间,仍然可以进行以下MDD的相关活动,例如产品变更及一年期内MDD新户的申请;现有MDD客户更新的相关活动;现有MDD客户更新的申请(包括提前更新的申请);现有MDD客户重大变更的申请期。虽然MDR的正式实施有延期,但是有例外情况如:
· MDD产品的投放截止日期仍为2024年5月26日
· MDD产品供应截止日期仍为2025年5月26日。
SUNGO可以在过渡期内为提供新旧法规的咨询服务,包括:
协助判定产品分类
协助选择合理的符合性途径
协助选择合适的认证机构
制定认证的解决方案
协助完成评估
编写CE技术文件
欧盟代表服务
http://sungofda.cn.b2b168.com