规格1
密度4
宽度2
质量5
高度3
制造商应计划在需要时尽快任命其英国负责人。然后,英国负责人必须根据上述宽限期,根据设备类别在MHRA中注册相关设备。
我公司办理产品出口欧盟、美国、中东南美等的各种认证: TUV莱茵,TUV南德等CE认证(MDD/MDR法规),CE技术文件编订, CE第四版评价(MEDDEV 2.7.1 Rev 4)编写,欧盟授权代表,欧盟自由销售证书,ISO13485:二016,器械单一审核程序(MDSAP)审核、美国FDA注册(含FDA510K申请),FDA美国代理人服务/ FDA QSR820验厂及整改,FDA警告信应对&RED LIST REMOVAL/QSR820体系/OTC验厂及整改,英国BRC认证咨询,BSCI验厂;口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试、防护服type5/6 (PPE认证咨询(89/686/EC个人防护指令)。 欧盟授权代表在欧盟的产品指令(Directive)英文版里使用的标准术语为(英国式英语) European Authorised Representative, 因为需要欧盟授权代表的多数为位于欧盟的的制造商,尤其以美国的制造商为多, 美国的制造商更喜欢将欧盟授权代表以美式英语书写为: European Authorized Representative。 鉴于中文并非欧盟的语言,因此European Authorised Representative 并没有对应的中文的术语。在中文的翻译里,通常将European Authorised Representative 或European Authorized Representative译为: 欧盟授权代表。 也有翻译为: 欧盟授权代理、欧洲授权代表、欧洲授权代理等。通常简称为:欧盟代表或欧代。也有使用:欧盟代理、欧洲代表、欧洲代理等。在闽台,还有使用欧体授权代表或欧体代表。 c) 欧盟制造商的产品在欧盟出现任何故障/事故/召回等问题,应由欧盟授权代表进行联络,通报,并与主管机关进行沟通联系。 如何选择一个的欧盟授权代表 a) 选择有资质,有能力的第三方欧盟授权代表公司。 欧盟境内注册的合法公司/拥有的技术人员,熟悉欧盟相关法规,能帮助制造方解决争端/避免空壳公司,代理商以及展会服务商。 ------以上几点可通过查询其欧盟注册证书,拨打其欧盟境内的电话看是否为电话录音等途径进行确认。 b) 签订有效的欧盟授权代表协议或合同。 合同/协议的甲乙双方的名称和地址必须和将来加贴CE标志的产品的包装/标签上的制造商和欧盟授权代表的名称和地址完全一致。 c) 应注意以下事项: 欧盟授权代表合同条款应是欧盟的主要语言版本,一般为欧代所在的语言版本。 避免在欧盟境内无固定办公地点和固定联络电话的欧盟授权代表。 避免经销商兼任欧盟授权代表。 避免在欧洲留学的亲戚朋友兼任欧盟授权代表。

在英国,必须在主管机构- 英国药物和健康产品管理局(MHRA)进行注册并取得注册证书和注册号码

当你次使用CE标志到您的设备上时,你必须通知主管机关进行注册(MHRA注册)
我公司办理产品出口欧盟、美国、中东南美等国家的各种认证:
TUV莱茵,TUV南德,等CE认证,CE技术文件编订, CE第四版评价(MEDDEV 2.7.1 Rev 4)编写,英国MHRA注册,欧盟授权代表,欧盟自由销售证书,ISO13485:2016,器械单一审核程序(MDSAP)审核、美国FDA注册(含FDA510K申请), FDA QSR820验厂及整改,FDA警告信应对&RED LIST REMOVAL/QSR820体系/OTC验厂及整改,英国BRC认证咨询,BSCI验厂;口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试、防护服type5/6 (PPE认证咨询(89/686/EC个人防护指令)。
谁应该向主管机关申请/注册?
欧洲经济区EEA的主管机关通常对器械的注册/通告收取一定费用。
对于一般器械:
你必须向你所注册的营业地点所在的EEA成员国的主管机关 (Competent Authority) 进行注册,如果你(属于下列任一):
制造 I类 器械或 定制式 (custom-made) 器械,并以你自己的公司名义或商标将其投放于EEA市场;
完全翻新I类器械,或为一个或多个现成的(I类)器械加贴标签,以期把它们以你自己的公司名(或商标)投放于EEA市场;
将市场上现有的带有CE标志的器械,在原制造商的预期使用目的和使用范围内,以你自己的公司名(或商标)置于一个系统或一个程序包之中;
为了能以你自己的公司名(或商标)投放于EEA市场,而对其他制造商制造的消毒后才可使用的系统或程序包或带有CE标志的器械进行消毒处理;
是位于欧洲经济区EEA的器械制造商的欧盟授权代表。
如果你是器械制造商但是在欧洲经济区EEA境内没有注册的营业地址的话,你必须在欧洲经济区EEA的会员国内有注册营业地址的一个授权代表来履行你的职责。
定制式 (custom-made) 器械是指根据一个的书写的而为某一个特定的病人使用非批量生产而制作的器械。
对于体外诊断器械(IVD):
你必须向你所注册的营业地点所在的EEA成员国的主管机关 (Competent Authority) 进行注册,如果你(属于下列任一):
制造体外诊断器械(IVD),并以你自己的公司名义或商标将其投放于EEA市场;
以你自己的公司名义或商标制造供性能评估用的体外诊断器械(IVD);
是一个在欧洲经济区EEA境内没有注册营业地址的体外诊断器械(IVD)制造商的欧盟授权代表。
如果你在欧洲经济区EEA境内没有注册的营业地址,而希望将体外诊断器械(IVD)投放EEA市场的话,你必须在欧洲经济区EEA的会员国内有注册营业地址的一个授权代表来履行你的职责。
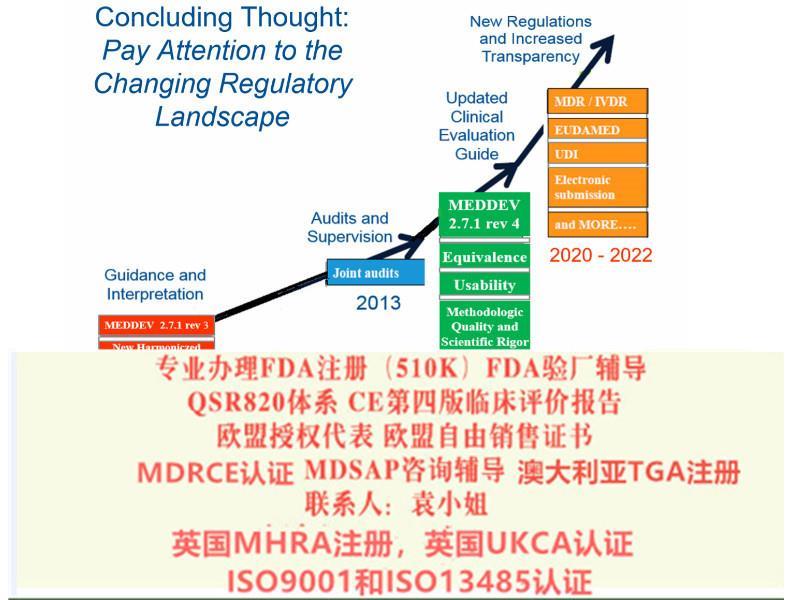
欧盟法规已经升级了,您的产品属于I类吗?欧盟新法规要求非常严苛,您是否按照新法规MDR/IVDR办理了CE了?是否有欧盟代表,欧盟注册,SRN号码,Basic UDI,是否已经申报数据库Eudamed?
http://sungofda.cn.b2b168.com










